
Chromosomal Engineering of Escherichia coli for Efficient Production of Coenzyme Q10*
2014-07-18 12:09:47HUANGMingtao黃明濤CHENYunyan陳韻妍andLIUJianzhong劉建忠BiotechnologyResearchCenterandMOEKeyLaboratoryofBioinorganicandSyntheticChemistrySchoolofLifeScienceSunYatSenUniversityGuangzhou510275China
HUANG Mingtao (黃明濤), CHEN Yunyan (陳韻妍) and LIU Jianzhong (劉建忠)**Biotechnology Research Center and MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Life Science, Sun Yat-Sen University, Guangzhou 510275, China
Chromosomal Engineering of Escherichia coli for Efficient Production of Coenzyme Q10*
HUANG Mingtao (黃明濤), CHEN Yunyan (陳韻妍) and LIU Jianzhong (劉建忠)**
Biotechnology Research Center and MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Life Science, Sun Yat-Sen University, Guangzhou 510275, China
The plasmid-expression system is routinely plagued by potential plasmid instability. Chromosomal integration is one powerful approach to overcome the problem. Herein we report a plasmid-free hyper-producer E. coli strain for coenzyme Q10production. A series of integration expression vectors, pxKC3T5b and pxKT5b, were constructed for chemically inducible chromosomal evolution (multiple copy integration) and replicon-free and markerless chromosomal integration (single copy integration), respectively. A coenzyme Q10hyper-producer Escherichia coli TBW20134 was constructed by applying chemically inducible chromosomal evolution, replicon-free and markerless chromosomal integration as well as deletion of menaquinone biosynthetic pathway. The engineered E. coli TBW20134 produced 10.7 mg per gram of dry cell mass (DCM) of coenzyme Q10when supplemented with 0.075 g·L?1of 4-hydroxy benzoic acid; this yield is unprecedented in E. coli and close to that of the commercial producer Agrobacterium tumefaciens. With this strain, the coenzyme Q10production capacity was very stable after 30 sequential transfers and no antibiotics were required during the fermentation process. The strategy presented may be useful as a general approach for construction of stable production strains synthesizing natural products where various copy numbers for different genes are concerned.
coenzyme Q10, Escherichia coli, chemically inducible chromosomal evolution, replicon-free and markerless chromosomal integration, chromosomal engineering
1 INTRODUCTION
Ubiquinone (coenzyme Q, abbreviation CoQ) is 2,3-dimethoxy,5-methyl,6-polyisoprene
para-benzoquinone and plays an essential role in the respiratory chain of many organisms. Coenzyme Q10(CoQ10) is a lipid-soluble antioxidant. It can be used as a nutritional supplement as well as in some cosmetics. Recently, CoQ10has been widely used as an orally administered prevention and therapy for a variety of diseases including cardiovascular diseases (such as congestive heart failure, ischemic heart disease and diastolic dysfunction of the left ventricle) and neurodegenerative diseases [1-4]. The reports on the potential health effects of CoQ10have resulted in an increase in demand for CoQ10and have led to the development of bioprocess for commercial production of CoQ10. Agrobacterium tumefaciens KCCM 10413 [5] and Rhodopseudomonas sphaeroides (a.k.a. Rhodobacter sphaeroides) [6, 7] were exclusively used in the commercial production of CoQ10. As the knowledge on biosynthetic enzymes and regulatory mechanism of CoQ production increased, synthesis of CoQ10by metabolic engineered microorganisms was constructed [8-20]. In our previous paper [21], E. coli TBW20108 was used to produce CoQ10. Plasmid-based co-overexpression of the pck, dxs, idi and ubiCA gene cluster and the ispA, ddsA and gapC gene cluster in E. coli TBW20108 produced CoQ10of 3.24 mg per gram of dry cell mass (DCM), which is unprecedented in E. coli. However, the content of CoQ10was lower than that with A. tumefaciens KCCM 10413 (11.84 mg·g?1DCM) [5], R. sphaeroides KY8598 (14.5 mg·g?1DCM) [6] and Sphingomonas sp. ZUTEO3 (32.5 mg·g?1DCM) [22], which were used in the commercial production of CoQ10. This indicates that further research is needed to increase CoQ10yields in E. coli.
However, all studies on the production of coenzyme Q10in E. coli use plasmids for the expression of a few key genes, which has some disadvantages. The recombinant vector may be structurally unstable, segregationally unstable or allele segregation in cells [23-25]. These plasmid instabilities cause genetic instability, reducing the production of the compound of interest. Apparently, these problems resulted from plasmids can be circumvented by chromosome engineering of the strain. Recently, two strategies for chromosomal engineering have been developed to overcome these disadvantages. The first strategy is called replicon-free and markerless methods (RMM) for chromosomal insertion of genes, developed by Chiang et al [26]. It is a single-copy expression system, which may be unsuitable for over-production of metabolite. The second is called chemically inducible chromosomal evolution (CIChE) developed by Tyo et al. to increase gene copy numbers for over-production of metabolite [27]. In this study, we apply the two methods to improve CoQ10production with E. coli. To our knowledge, this is the first report on engineering application of a plasmid-free E. coli for enhancing CoQ10production by applying CIChE and RMM.
2 MATERIALS AND METHODS
2.1 Strains, primers and plasmids
Strains, primers and plasmids used in this study are listed in Table 1. E. coli DH5α was used for plasmid construction. E. coli TBW20108 was used as the parent strain for chromosomal integration. CoQ10was purchased from Sigma.
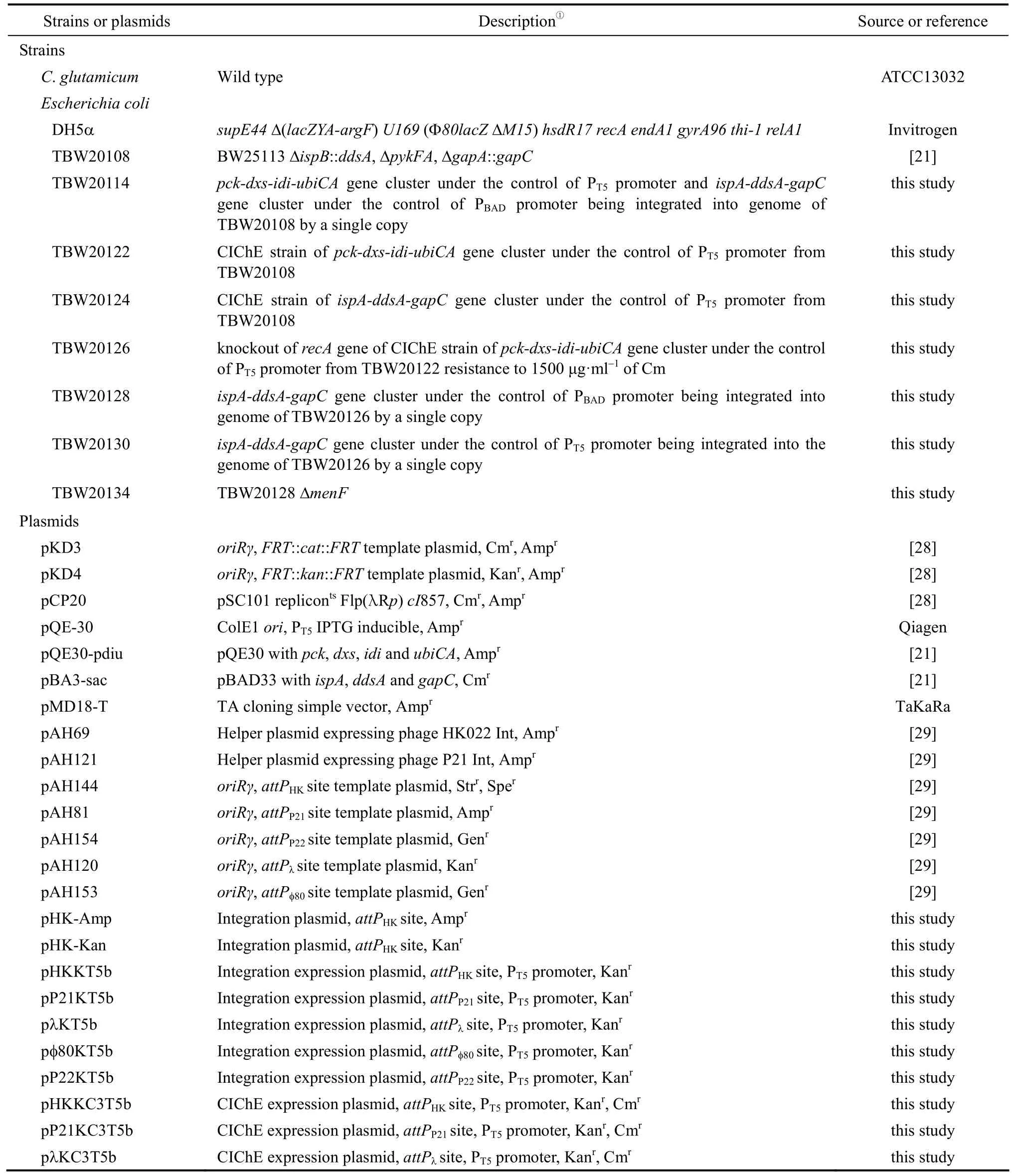
Table 1 Strains, plasmids and primers used in this study
2.2 Integration expression plasmid construction
Because the plasmids developed by Chiang et al.
have not any promoter in front of multiple cloning site (MCS) and the integration using these plasmids needs two rounds of PCR [26], we modified the integration plasmid for RMM by the insertion of T5 promoter and transcription terminator to obtain a series of RMM integration expression vectors pxKT5b. The DNA fragment containing MCS and the attP site from pAH144 was amplified with PstI and XhoI restriction site overhangs using primers RCWF and RCWR. The fragment containing the ampicillin marker and the origin of replication from pKD3 was amplified with PstI and XhoI restriction site overhangs using primers RCNF and RCNR. The two fragments were digested and ligated together to form pHK-Amp. The bla gene (encoding β-lactamase) in pHK-Amp was cut out by SphI and NotI and replaced with the kan gene (encoding aminoglycoside 3′-phosphotransferase) from pAH120 to produce pHK-Kan. The cat gene (encoding chloramphenicol acetyltransferase) from pKD3 was amplified using primers KI3 and KI4. The PCR product was digested with EcoT221 and MunI and ligated into the PstI/EcoRI sites of pHK-Kan to form pHKK-KI3. Plasmid pHKK-KI3 was then cut with SphI, treated with Primestar HS DNA polymerase [TaKaRa Biotechnology (Dalian) Co., Ltd., China] and recircularized by ligation to form pHKK-KI3b. This resulted in the elimination of SphI site. The DNA fragment containing T5 promoter, MCS and terminator from pQE30 was amplified by PCR using primers Q3 and Q4, and ligated into pMD18-T simple vector to form p18S-Q3Fb. The PCR fragment was digested along with the pHHK-KI3b backbone using MluI and ApaLI and ligated together to form pHKKT5b. The DNA fragments containing various attP sites (attPP21for phage P21, attPφ80for phage φ80, attPλfor phage λ and attPP22for phage P22) were amplified by PCR using primers AH2 and AH3 from pAH81, pAH153, pAH120 and pAH154 and ligated into pMD18-T simple vector to form pMDS-P21, pMDS- φ, pMDS-λ and pMDS-P22, respectively. The fragments were digested along with the pHKKT5b backbone using ApaLI and XhoI and ligated together to form pP21KT5b, pφ80KT5b, pλKT5b and pP22KT5b, respectively. These vectors can be used to integrate a gene of interest into a chromosome by direct transformation for RMM. The maps of these vectors are presented in Fig. 1 (a). We can use these vectors to insert genes of interest into the chromosome by one-time PCR.

Table 1 (Continued)
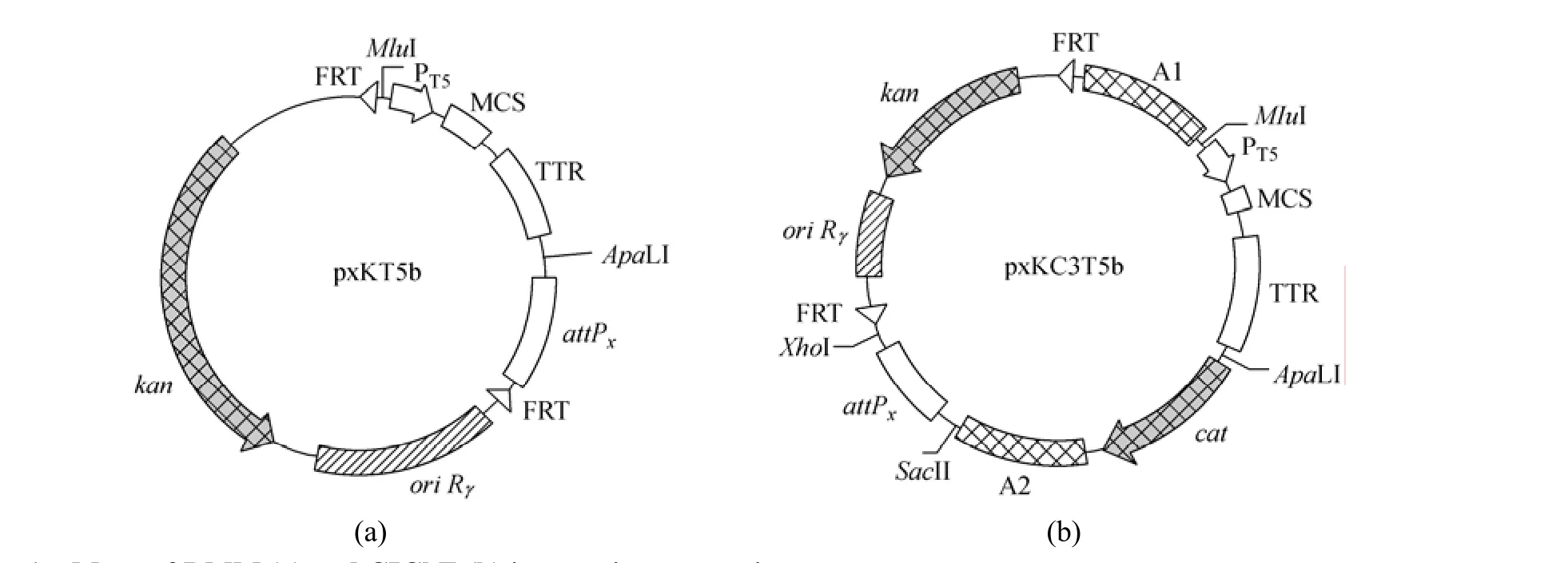
Figure 1 Maps of RMM (a) and CIChE (b) integration expression vectors [x: HK for phage HK022, P21 for phage P21, φ80 for phage φ80, λ for phage λ or P22 for phage P22; MCS: EcoRI, BamHI, SphI, SacI, KpnI, SmaI, SalI and PstI; PT5: T5 promoter from pQE30; TTR: terminator from pQE30; ori Rγ: γ replication origin of R6K requiring trans-acting π protein (pir gene product) for replication; FRT: FLP recognition target; kan: aminoglycoside 3′-phosphotransferase for kanamycin resistance; cat: chloramphenicol acetyltransferase for chloramphenicol resistance; A1 and A2: non-coding 1 kb region of cgl1740 gene from C. glutamicum ATCC 13032]
For CIChE, Tyo et al. constructed an integration plasmid (pTGD) that uses the λInCh integration system for delivery to the genome [27]. The λInCh genomic integration protocol is more complicated and time-consuming, because it contains three steps that involve two recombination steps [31]. To overcome the drawbacks of CIChE originally devised by Tyo et al., we constructed a series of CIChE integration expression vectors [Fig. 1 (b)] based on our RMM integration expression vectors. The cgl1740 gene of C. glutamicum ATCC 13032 was amplified using primers CG1F and CG1R, and cloned into the PstI site of pHK-Kan to form pHK-KanA1. The cgl1740 gene of C. glutamicum ATCC 13032 was also amplified using primers CG2F and CG2R, and cloned into the EcoRI site of pHK-KanA1 to form pHK-KanA2. The cgl1740 gene in pHK-KanA2 was used as the identical, and non-coding 1 kb region as the chlB gene in pTGD [27]. The cat gene from pKD3 was amplified by PCR using primers CM1 and CM2, and cloned into the EcoRI site of pHK-KanA2 to form pHK-KanC3. Plasmid pHK-KanC3 was then cut with SphI and treated with Primestar HS DNA polymerase and recircularized by ligation to form pHK-KanC3b. This resulted in the elimination of SphI site. The DNA fragment containing T5 promoter, MCS and terminator was excised using MluI and ApaLI from p18S-Q3Fb and cloned into the MluI/ApaLI sites of pHK-KanC3b to form pHKKC3T5b. The DNA fragments containing various attP sites (attPP21for phage P21, attPφ80for phageφ80, attPλfor phage λ and attPP22for phage P22) were amplified by PCR using primers AH1 and AH2 from pAH81, pAH153, pAH120 and pAH154 and ligated into pMD18-T simple vector to form p18S-P21, p18S-φ, p18S-λ and p18S-P22, respectively. The fragments were digested along with pHKKC3T5b backbone using SacII and XhoI and ligated together to form pP21KC3T5b, pφ80KC3T5b, pλKC3T5b and pP22KC3T5b, respectively. These vectors can be used to integrate a target gene into a chromosome by direct transformation and then to carry out CIChE. The maps of these vectors are presented in Fig. 1 (b).
2.3 Chromosomal integration
The ispA, ddsA and gapC gene cluster under the control of PBADfrom the pBA3sac plasmid was cut out using the restriction enzymes MluI and SphI and then cloned into the MluI/SphI sites of pHKKT5b to form the pHKKb-BAsac integration vector. The pck-dxs-idiubiCA gene cluster from the pQE30-pdiu plasmid was cut out using the restriction enzymes EcoRI and PstI and then cloned into the EcoRI/PstI sites of pHKKT5b to form the pHKKT5b-pdiu integration vector. The resulting integration vector was inserted into the bacterial attachment (attB) site of E. coli as a single copy through the use of a helper plasmid pAH69 expressing the phage integrase (Int) by direct transformation as described by Chiang et al. [26] and Haldimann and Wanner [29]. The general procedure for using these integration vectors was illustrated in Fig. 2 (a). In brief, strains containing the helper plasmid pAH69 were first cultured in SOB medium (super optimal broth) with ampicillin (Amp) at 30 °C to an optical density at 600 nm of ca. 0.6. The cells were made electrocompetent and transformed with the integration vector pHKKb-BAsac. Following electroporation, cells were suspended in SOC (super optimal broth with catabolic repressor) without Amp, incubated at 37 °C for 1 h and at 42 °C for 30 min, and then spread onto LB (Luria Bertani) agar plates containing kanamycin (Kan) and incubated overnight at 37 °C. Colonies were verified by colony PCR using primer sets of HKP1 and AHP2, or AHP3 and HKP4. The correct colony was cultured in 5 ml LB medium with Kan at 42 °C for overnight and then spread onto LB medium with Kan agar plate at 37 °C for overnight and finally tested for stable integration by antibiotic resistance (conferring Kan resistance) and loss of the helper plasmid (conferring Amp sensitivity) as well as by colony PCR using primer sets of HKP1 and AHP2, or AHP3 and HKP4. To eliminate the region containing the selective marker and the replication origin, the resulting integrants bearing the inserted DNA were transformed with pCP20 expressing FLP [28]. Colonies were verified by colony PCR using primer sets of HKP1 and PAR, or AHP3 and HKP4.
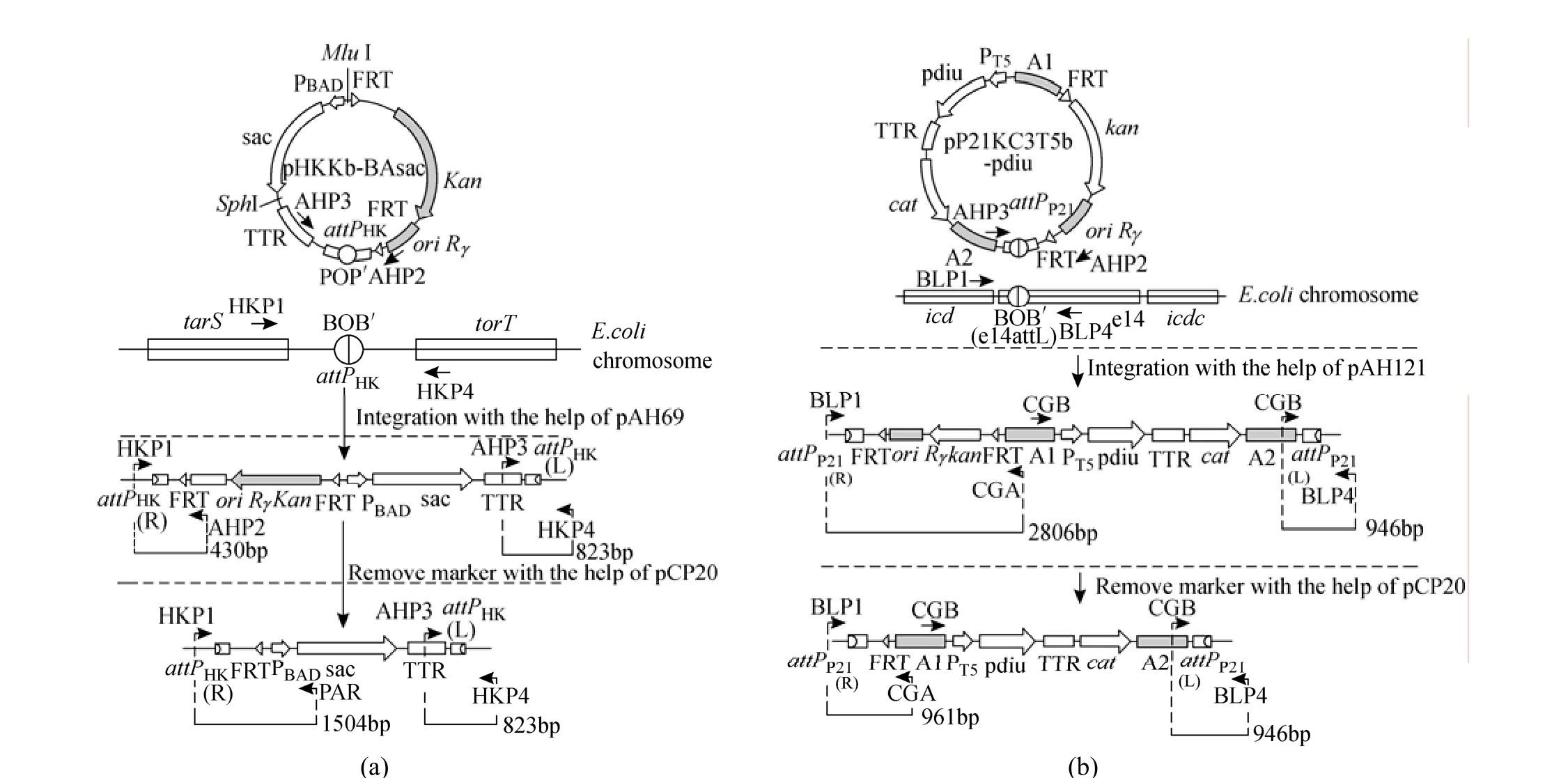
Figure 2 The change of genomic structure by integration of pHKKb-BAsac at attPHKsite (a) or by integration of pP21KC3T5-pdiu at attPP21site (b) [with primers HKP1, AHP2, AHP3, HKP4, PAR, BLP1, CGA, CGB, BLP4 (Table 1) used for verification by colony PCR; the numbers beside bars: expected size of PCR fragments amplified using corresponding primer sets (shown by arrows)]
2.4 Chemically inducible chromosomal evolution
The gene cluster containing pck, dxs, idi and ubiCA genes was excised from the pQE30-pdiu plasmidby EcoRI and PstI and cloned into the EcoRI/PstI sites of pP21KC3T5b to form pP21KC3T5b-pdiu. Similarly, pP21KC3b-BAsac plasmid was constructed by digesting the ispA, ddsA and gapC gene cluster under the control of PBADfrom the pBA3sac plasmid with MluI and SphI and then cloning it into the MluI/SphI sites of pP21KC3T5b to form pP21KC3T5b-BAsac. The resulting DNA constructs were integrated into the chromosome of E. coli TBW 20108 as described above. The general procedure for using these integration vectors was illustrated in Fig. 2 (b). Colonies were verified by colony PCR using primers in Fig. 2 (b).
The CIChE of the above construct was carried out by subculturing the resulting strains in 5 ml SOB medium with increasing concentrations of chloramphenicol in 15 ml culture tubes as described by Tyo et al [27]. The strain was grown to stationary phase in 20 μg·ml?1chloramphenicol. 50 μl of the culture was subcultured in a new culture tube, in which the chloramphenicol concentration was doubled from 20 μg·ml?1to 40 μg·ml?1and allowed to grow to stationary phase. The process was repeated until the desired concentration was reached. The recA gene of CIChE strain was then deleted (see below).
2.5 Gene knockout
Knockouts of recA and menF genes were carried out by PCR product recombination [28] using the pSIM6 plasmid [30] expressing the λ red recombination system and pKD4 as the template for PCR. Gene knockouts were verified using colony PCR using primers VCA1 and VCA2 for the deletion of recA gene, VNF1 and VNF2 for the deletion of menF gene.
2.6 qPCR measurement of gene copy number
Gene copy numbers were measured by qPCR on genomic DNA isolated from appropriate CIChE strains. qPCR was performed with an iCycler iQ5 Real Time PCR system (Bio-Rad Laboratories, USA) using the SYBR Green Premix Ex Taq (Bio-Rad). The copy numbers of the cat gene were detected and compared to the copy number of minD, a nearby native gene in the chromosome. The primers QCF and QCF were used to measure the copy number of cat. The primers NDF and NDR were used to measure the copy number of minD.
2.7 CoQ10production
E. coli was grown in LB broth or LB agar. SOB and SOC media were used to prepare electrocompetent cells and for recovery after transformation. Appropriate antibiotics (Amp 100 μg·ml?1, Kan 25 μg·ml?1and Cm 25 μg·ml?1) were added to the culture media when needed. As for CoQ10production, 5 ml of LB medium was used for overnight pre-cultivation of E. coli in a test tube at 37 °C. Main cultures were grown
in 250 ml flasks containing 25 ml SOB medium with phosphate salt, glycerol and 4-hydroxy benzoic acid (SPGB). SPGB medium [21] contains (g·L?1): peptone 20, yeast extract 5, NaCl 0.5, KCl 0.186, MgCl2?6H2O 0.95, KH2PO43.0, Na2HPO4·12H2O 17.1, glycerol 20, and 4-hydroxy benzoic acid (4HB) 0.075. Main cultures were inoculated with a starting OD600of 0.1 and incubated at 37 °C for 24 h in a rotary shaking incubator at 200 r·min?1. For characterization of CoQ10production, batch fermentation was performed in shaking flask as above. Samples were taken and measured with an interval of 4 h.
2.8 Assay
Cell growth was measured by optical density at 600 nm and converted into dry cell mass (g DCM per liter) using a standard curve. Cells, cultured at 37°C for 24 h, were collected from 20 ml medium by centrifugation, then washed once with water and stored at?20 °C before extraction. The cell pellet was suspended in 25 ml acetone and broken by sonication for 8 min in an ultrasonic disintegrator. Then 20 ml petroleum ether was added to the fragmentation and shaken vigorously. Cell debris was removed by centrifugation at 7000 g for 10 min. The solvent phase was filtrated and evaporated to dryness in a vacuum. The residue was dissolved in ethanol. CoQ10was quantified by a HP1100 HPLC system (Hewlett Packard, USA) equipped with a Diamondsil C18column (250 mm×4.6 mm, 5 μm, Dikma Co., Beijing, China). 100% ethanol was used as a mobile phase at a flow rate of 0.8 ml·min?1. A UV detector was used at 275 nm for the detection and quantification of CoQ10. A standard curve was generated for the quantification of CoQ10by making serial dilutions of a CoQ10standard stock solution (1 mg·ml?1). The glycerol concentration of the fermentation broth was measured by a colorimetric method using Nash reagent [32].
OriginPro (version 7.5) package was used to perform ANOVA and pairwise comparison according to Tukey’s test. Statistical significance was inferred at a P value of 0.05.
3 RESULTS AND DISCUSSION
3.1 Integration expression plasmid construction
The RMM integration expression vectors constructed in this study can be integrated into the chromosome by direct transformation with the aid of appropriate helper plasmid and then the replication origin and the antibiotic-resistant marker in the integration expression vector can be eliminated with the help of pCP20 [Fig. 2 (a)]. The resulting strain is a “clear”producer.
The CIChE integration expression vectors constructed in this study can deliver a duplication-ready expression cassette into the E. coli chromosome by direct transformation [Fig. 2 (b)]. These vectors alsohave an MCS and the antibiotic-resistant gene (cat) flanked on both sides by the homologous regions as pTGD [27] and can be used for chromosomal evolution. After integrating genes of interest, the chromosome will evolve to possess higher gene copy numbers with the help of recA-dependent homologous recombination by subculturing the strain in increasing concentrations of Cm. At the desired gene copy number, recA can be deleted to prevent further change in copy number. Thus the protocol of gene integration and chromosomal evolution using our CIChE integration expression vectors is simpler and more efficient than that using the vector of Tyo et al [27].
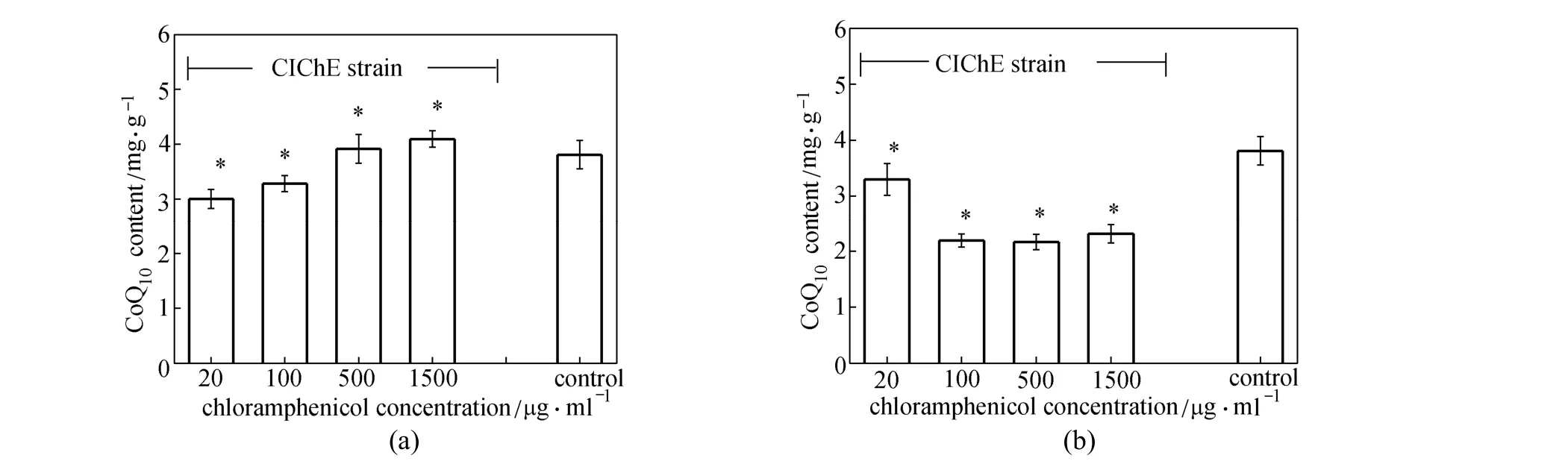
Figure 3 CoQ10production by CIChE strains under different Cm concentrations (a) E. coli TBW20122 harboring pBA3sac; (b) E. coli TBW20124 harboring pQE30-pdiu; with E. coli TBW20108 harboring pQE30-pdiu and pBA3sac as a control, cells cultured in SPGB medium at 37°C for 24 h, 0.2% arabinose and 0.5 mmol·L?1IPTG added to all media for induction; data: mean ± SD for three replicates; *: significant different at P<0.05 compared to corresponding controls
3.2 Chromosomal engineering
In our previous study [21], a deleted E. coli TBW20108 was constructed for CoQ10production. Co-overexpression of the gene clusters (pck-dxs-idiubiCA and ispA-ddsA-gapC) in E. coli TBW20108 enhanced CoQ10production. In order to overcome the drawbacks of using a plasmid-expression system, we first applied CIChE for the above two gene clusters (pck-dxs-idi-ubiCA and ispA-ddsA-gapC) to obtain CIChE strains E. coli TBW20122 and TBW20124, respectively. Fig. 3 shows the results of CoQ10production of CIChE strains containing appropriate plasmid (pBA3sac for CIChE strains E. coli TBW20122 or pQE30-pdiu for CIChE strains E. coli TBW20124) and E. coli TBW20108 (pQE30-pdiu, pBA3sac). From Fig. 3 (a), we can see that CoQ10production in CIChE strains of the pck-dxs-idi-ubiCA gene cluster (E. coli TBW20122) along with plasmid-based overexpression of the ispA, ddsA and gapC gene cluster increases as more chloramphenicol (Cm) is used in chromosomal evolution, with the maximal CoQ10production of (4.1±0.2) mg·g?1DCM by E. coli TBW20122 resistance to 1500 μg·ml?1of Cm bearing pBA3sac. The CoQ10production in the CIChE strain did not increase when the Cm concentration was above 1500 μg·ml?1in chromosomal evolution (data not shown). From Fig. 3 (b), we can see that CoQ10production of CIChE strains of the ispA-ddsA-gapC gene cluster (E. coli TBW20124) harboring pQE30-pdiu decreases as more Cm is used in chromosomal evolution. This result indicates that the ispA-ddsA-gapC gene cluster is not suitable for chromosomal evolution.
In order to investigate the relationship between copy numbers of the integrated gene cluster and CoQ10production, gene copy numbers of CIChE strains were measured (Fig. 4). Gene copy numbers of CIChE strains increased as more Cm was used in chromosomal evolution. The gene copy numbers in the CIChE strain of the ispA-ddsA-gapC gene cluster were higher than that of the pck-dxs-idi-ubiCA gene cluster. It may be that the nucleotide length of the pck-dxs-idi-ubiCA gene cluster (ca. 5600 bp) is longer than that of the ispA-ddsA-gapC gene cluster (ca. 3600 bp). Tyo et al. reported a similar result. They found that the CIChE strain without a polyhydroxybutyrate (PHB) operon had higher copy numbers than that with a PHB operon [27].
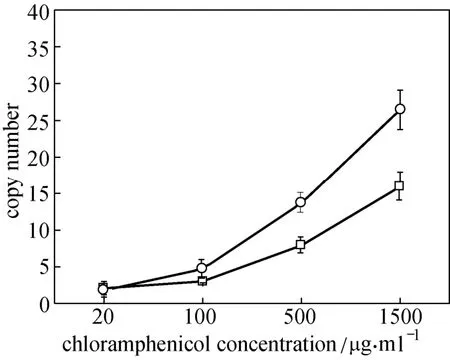
Figure 4 Gene copy numbers of integrated gene cluster in CIChE strains at different Cm concentrations□ CIChE strains of pck-dxs-idi-ubiCA gene cluster (TBW20122);○ CIChE strains of ispA-ddsA-gapC gene cluster (TBW20124); data: mean ± SD for three replicates
From Fig. 3, we can also find that CoQ10productions of the CIChE strains of the pck-dxs-idi-ubiCA gene cluster at lower Cm concentration (20-100 mg·ml?1)were lower than that of the control strain (E. coli TBW 20108 harboring pQE30-pdiu and pBA3-sac), since the copy number (2-3) of the pck-dxs-idi-ubiCA gene in the CIChE strains (E. coli TBW20122) at lower Cm concentration may be lower than that of the control strain.
Figures 3 and 4 also show that higher copy numbers of the gene cluster of pck-dxs-idi-ubiCA and lower copy numbers of the gene cluster of ispA-ddsA-gapC are beneficial to CoQ10production in E. coli. This indicates that the availability of isoprenoid precursors (isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP)) and the prenylation of 4-hydroxy benzoic acid (4HB) are more important for CoQ10biosynthesis in E. coli. The over-expression of pck, dxs and idi genes increases the production of isoprenoid precursors for CoQ10biosynthesis. This point has been proved by other groups. Over-expression of foreign mevalonate pathway can significantly improve the availability of isoprenoid precursors, increasing CoQ10content in engineered E. coli significantly [8, 13].
Thus, we deleted the recA gene of the CIChE strain of pck-dxs-idi-ubiCA at a Cm concentration of 1500 μg·ml?1to prevent subsequent homologous recombination to obtain E. coli TBW20126. We subsequently inserted a single copy of the gene cluster of ispA-ddsA-gapC using RMM into the genome of E. coli TBW20126 to obtain E. coli TBW20128 and then assayed for CoQ10production (Fig. 5). E. coli TBW20128 produced (4.7±0.2) mg·g?1DCM of CoQ10, higher than that (3.8±0.3) mg·g?1DCM of E. coli TBW20108 (pQE30-pdiu, pBA3-sac). In order to investigate the effect of copy numbers of the gene cluster of pck-dxs-idi-ubiCA, we also inserted a single copy of the gene cluster of pck-dxs-idi-ubiCA and ispA-ddsA-gapC using RMM into the genome of E. coli TBW20108 to obtain E. coli TBW20114. E. coli TBW20114 produced (1.8±0.1) mg·g?1DCM of CoQ10, significantly lower than that of E. coli TBW20128 (Fig. 5). We also replaced the promoter of the ispA-ddsA-gapC gene cluster (PBAD) with stronger promoter (PT5) to obtain E. coli TBW20130 to assess the effect of promoter. The difference in the levels of CoQ10production between E. coli TBW20128 and TBW20130 was not significant (Fig. 5).
Figure 5 shows that the level of CoQ10production in E. coli TBW20114, TBW20128 and TBW20130 remained a constant after 10 rounds of subculture without the antibiotic Cm, even after 30 rounds (data not shown). However, the level of CoQ10production in E. coli TBW20108 (pQE30-pdiu, pBA3-sac) sharply decreased after 10 rounds of subculture in the presence of appropriate antibiotics. These results are consistent with previous reports wherein the gene copy number and PHB production of CIChE strain remained constant after 40 rounds of subculture [27]. It was also found that PHB productivity of the plasmid-carrying strain cultured with antibiotics was completely lost after 35 generations. The plasmid system with antibiotics lost productivity may be due to allele segregation. It is likely to occur regularly, despite using antibiotic selection, particularly in subculturing experiments or chemostats [27].
In E. coli, chorismate, which is the precursor for the biosynthesis of CoQ, can be converted to isochorismate to form menaquinone (vitamin K) by isochorismate synthase, which is encoded by the menF gene under anaerobic conditions [33, 34]. Thus, we deleted the menF gene of E. coli TBW20128 to obtain E. coli TBW20134 and assayed for CoQ10production. E. coli TBW20134 produced CoQ10up to (5.2±0.3) mg·g?1DCM after 24 h.
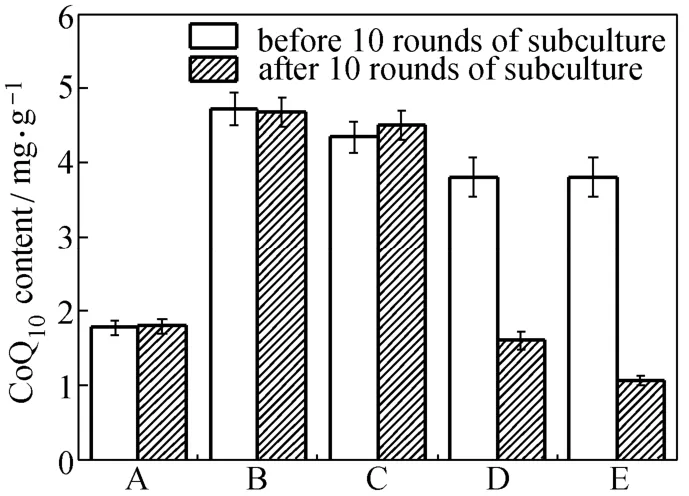
Figure 5 CoQ10production and genetic stability of the engineered strains and the two-plasmid-bearing strain (A) E. coli TBW20114; (B) E. coli TBW20130; (C) E. coli TBW20128; (D) E. coli TBW20108 harboring pQE30-pdiu and pBA3sac with appropriate antibiotic in successive subculture; (E) E. coli TBW20108 harboring pQE30-pdiu and pBA3sac without antibiotic in successive subculture; with cells cultured in SPGB medium at 37 °C for 24 h, 0.2% arabinose and 0.5 mmol·L?1IPTG added in all media for induction, and no antibiotics used unless specifically mentioned; data: mean ± SD for three replicates
3.3 Characterization of CoQ10production
For a more detailed view on CoQ10production, batch culture of E. coli TBW20134 was carried out in shaking flasks. The time profiles of cell growth, glycerol consumption and CoQ10production are presented in Fig. 6. The growth of the engineered strain entered the exponential growth phase fast. After 44 h, the glycerol concentration decreased to below 1.0 g·L?1. During the exponential growth phase, CoQ10production increased exponentially as expected. After transition to the stationary growth phase, CoQ10production still increased. The maximal CoQ10content of (10.7±0.6) mg·g?1DCM and CoQ10concentration of (77.4±4.2) mg·L?1were obtained at 72 h when supplemented with 4HB of 0.075 g·L?1. Among natural producers, A. tumefaciens and R. sphaeroides were identified as good candidates for CoQ10production [21, 35, 36]. Ha et al. reported that the highest specific CoQ10production of 11.84 mg·g?1DCM was obtained in a pH-stat fed-batch culture using A. tumefaciens [5]. Sakato et al. reported the highest value (12.5 mg·g?1DCM) of specific CoQ10production in Rhodopseudomonas spheroids KY-8598 by fed-batch cultivation in 30 L bioreactor [6]. However, there has not been a scaled-up study using Rhodopseudomonas spheroidsKY-8598. Kien et al. showed that R. sphaeroides produced 8.12 mg·g?1DCM of CoQ10by fed-batch fermentation in 150 L bioreactor [37]. The coupled fermentation-extraction process led Sphingomonas sp. ZUTEO3 to produce CoQ10of 32.5 mg·g?1DCM [22], which may be one potential CoQ10production strategy with higher yield and lower cost. The highest yield of CoQ10produced in E. coli (2.43 mg·g?1DCM) with supplementation of 0.01% 4HB after 24 h was reported by Zahiri et al. [13], while our previous paper reported that a recombinant E. coli harboring two plasmids produced 3.24 mg·g?1DCM of CoQ10[20]. In this study, we constructed an engineered E. coli TBW20134 strain that produced (10.7±0.6) mg·g?1DCM of CoQ10with 4HB of 0.075 g·L?1supplemented and antibiotics-less. Even with the same culture time (24 h), the yield [(5.2±0.3) mg·g?1DCM] of CoQ10of E. coli TBW20134 was also higher than that reported by Zahiri et al [13]. The specific CoQ10production of E. coli TBW20134 is close to the level of A. tumefaciens and the highest in E. coli so far. Thus, our engineered E. coli TBW20134 may be a good candidate for CoQ10production. The yield may be enhanced by optimization of culture conditions and fed-batch fermentation. These are currently underway in our lab and the results will be reported shortly.
4HB is the aromatic precursor to CoQn. Zahiri et al. reported that addition of 4HB significantly improved CoQ10production in engineered E. coli [13]. In this study, all the above data were also obtained by 4HB supplementation. To investigate the effect of 4HB supplementation on CoQ10production, E. coli TBW20134 and TBW20128 were cultured in the main medium without 4HB. The results are presented in Fig. 7. Supplementation of 4HB significantly improves CoQ10production for the two strains. E. coli TBW20134 without 4HB can only produce CoQ10of (6.1±0.2) mg·g?1DCM after 72 h. The value is much lower than that [(10.7±0.6) mg·g?1DCM] with 4HB supplementation, but it is about 2.5-fold that (2.43 mg·g?1DCM) reported by Zahiri et al [13]. Cluis et al. reported that increasing the endogenous availability of 4HB by over-expressing genes coding for rate-limiting enzymes of the aromatic pathway significantly enhanced CoQ10production in E. coli [8]. Thus, increasing endogenous availability of aromatic precursor in E. coli TBW20134 may further improve CoQ10production.
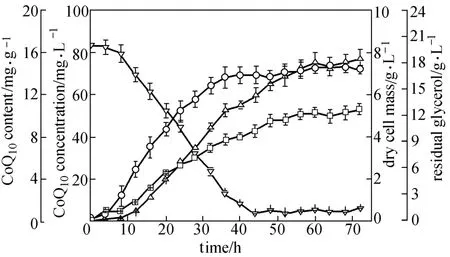
Figure 6 Time course of CoQ10batch fermentation by E. coli TBW20134 in SPGB medium containing 0.2% arabinose and 0.5 mmol·L?1IPTG at 37 °C and 200 r·min?1□ CoQ10content; △ CoQ10concentration; ○ dry cell mass; ▽ residual glycerol; data: mean ± SD for three replicates
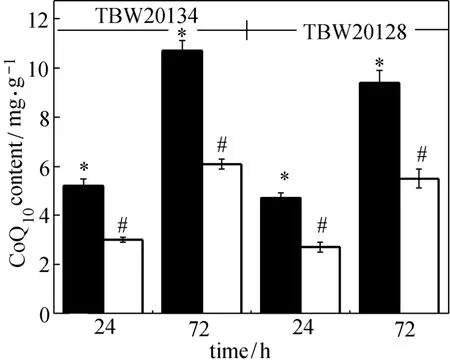
Figure 7 Effects of 4HB supplementation on CoQ10production by E. coli TBW20134 and TBW 20128 filled bar: 4HB supplementation; open bar: without 4HB supplementation; * and #: statistically significant at P<0.05
To determine the nature of CoQ species, CoQ, extracted from E. coli TBW20134 cultured in shake flask after 72 h, was analyzed by HPLC. CoQ10was detected as the main product with a small amount of CoQ8and CoQ9(less than 1%). The result is consistent with that obtained by replacement E. coli of the ispB gene [8, 12]. We did not observe detectable amounts of menaquinone (MK-10) either. However, E. coli CC2.D produced MK-10 [8].
Although the yield of our engineered E. coli TBW20134 without supplementation of antibiotics is close to that of the commercial producer A. tumefaciens, it has multiple copy of cat gene which is a chloramphenicol resistance marker. There is a potential safe problem for the spread of the antibiotic-resistant trait to other microbes in nature. Replacement of the cat gene in these integration expression vectors with the growth essential target gene fabI may be a good strategy to avoid the spread of the antibiotic resistant marker [38]. Because CIChE is a high gene copy expression system, our engineered strain also has metabolic burden problem caused by the superfluous DNA sequences (the cat gene and the homologous region of 1 kb). Promoter engineering may be an alternativeapproach for gene over-expression.
4 CONCLUSIONS
In this study, we constructed a series of integration expression vectors for single copy or multiple copy integration, which can be applied in RMM and CIChE. We engineered a plasmid-free E. coli TBW20134, using CIChE, RMM as well as deletion of menaquinone biosynthetic pathway. The engineered E. coli TBW20134 produced CoQ10content of (10.7±0.6) mg·g?1DCM and CoQ10concentration of (77.4±4.2) mg·L?1. Moreover, no antibiotics were used in the fermentation process for the strain. This can reduce the cost and avoid the problem caused by addition of antibiotics. The engineered strain remained stable as determined by examination of its CoQ10-producing capacity after 30 sequential transfers. This study demonstrated that applying CIChE along with RMM could flexibly control gene copy numbers. The proposed methods can be applied to the production of other metabolites where various copy numbers for different genes is concerned.
REFERENCES
1 Bhagavan, H.N., Chopra, R.K., “Potential role of ubiquinone (coenzyme Q10) in pediatric cardiomyopathy”, Clin. Nutr., 24, 331-338 (2005).
2 Matthews, R.T., Yang, L., Browne, S., Baik, M., Beal, M.F., “Coenzyme Q10administration increases brain mitochondrial concentrations and exerts neuroprotective effects”, Proc. Natl. Acad. Sci., 95, 8892-8897 (1998).
3 Rosenfeldt, F., Marasco, S., Lyon, W., Wowk, M., Sheeran, F., Bailey, M., Esmore, D., Davis, B., Pick, A., Rabinov, A., Smith, J., Nagley, P., Pepe, S., “Coenzyme Q10therapy before cardiac surgery improves mitochondrial function and in vitro contractility of myocardial tissue”, J. Thorac. Cardiovasc. Surg., 129, 25-32 (2005).
4 Tran, M.T., Pharm, D., Mitchell, T.M., Kennedy, D.T., Giles, J.T.,“Role of coenzyme Q10in chronic heart failure, angina, and hypertension”, Pharmacotherapy, 21, 797-806 (2001).
5 Ha, S.J., Kim, S.Y., Seo, J.H., Jeya, M., Zhang, Y.W., Ramu, T., Kim, I.W., Lee, J.K., “Ca2+increases the specific coenzyme Q10content in Agrobacterium tumefaciens”, Bioprocess Biosyst. Eng., 32, 697-700 (2009).
6 Sakato, K., Tanaka, H., Shibata, S., Kuratsu, Y., “Agitation-aeration studies on coenzyme Q10production using Rhodopseudomonas spheroids”, Biotechnol. Appl. Biochem., 16, 19-22 (1992).
7 Yoshida, H., Kotani, Y., Ochiai, K., Araki, K., “Production of ubiquinone-10 using bacteria”, J. Gen. Appl. Microbiol., 44, 19-26 (1998).
8 Cluis, C.P., Ekins, A., Narcross, L., Jiang, H., Gold, N.D., Burja, A.M., Martin, V.J.J., “Identification of bottlenecks in Escherichia coli engineered for the production of CoQ10”, Metab. Eng., 13, 733-744 (2011).
9 Lee, J.K., Her, G., Kim, S.Y., Seo, J.H., “Cloning and functional expression of the dps gene encoding decaprenyl diphosphate synthase from Agrobacterium tumefaciens”, Biotechnol. Prog., 20, 51-56 (2004).
10 Lee, J.K., Oh, D.K., Kim, S.Y., “Cloning and characterization of the dxs gene, encoding 1-deoxy-d-xylulose 5-phosphate synthase from Agrobacterium tumefaciens, and its overexpression in Agrobacterium tumefaciens”, J. Biotechnol., 128, 555-566 (2007).
11 Park, Y.C., Kim, S.J., Choi, J.H., Lee, W.H., Park, K.M., Kawamukai, M., Ryu, Y.W., Seo, J.H., “Batch and fed-batch production of coenzyme Q10in recombinant Escherichia coli containing the decaprenyl diphosphate synthase gene from Gluconobacter suboxydans”, Appl. Microbiol. Biotechnol., 67, 192-196 (2005).
12 Zahiri, H.S., Noghabi, K.A., Shin, Y.C., “Biochemical characterization of the decaprenyl diphosphate synthase of Rhodobacter sphaeroides for coenzyme Q10production”, Appl. Microbiol. Biotechnol., 73, 796-806 (2006).
13 Zahiri, H.S., Yoon, S.H., Keasling, J.D., Lee, S.H., Kim, S.W., Yoon, S.C., Shin, Y.C., “Coenzyme Q10production in recombinant Escherichia coli strains engineered with a heterologous decaprenyl diphosphate synthase gene and foreign mevalonate pathway”, Metab. Eng., 8, 406-416 (2006).
14 Kim, S.J., Kim, M.D., Choi, J.H., Kim, S.Y., Ryu, Y.W., Seo, J.H.,“Amplification of 1-deoxy-D-xyluose 5-phosphate (DXP) synthase level increases coenzyme Q10production in recombinant Escherichia coli”, Appl. Microbiol. Biotechnol., 72, 982-985 (2006).
15 Kwon, O., Druce-Hoffman, M. Meganathan, R., “Regulation of the ubiquinone (coenzyme Q) biosynthetic genes ubiCA in Escherichia coli”, Curr. Microbiol., 50, 180-189 (2005).
16 Zhang, D., Li, Z., Wang, F., Shrestha, B., Tian, P., Tan, T., “Expression of various genes to enhance ubiquinone metabolic pathway in Agrobacterium tumefaciens”, Enzyme Microb. Technol., 41, 772-779 (2007).
17 Zhu, X., Yuasa, M., Okada, K., Suzuki, K., Nakagawa, T., Kawamukai, M., Matsuda, H., “Production of ubiquinone in Escherichia coli by expression of various genes responsible for ubiquinone biosynthesis”, J. Ferment. Bioeng., 79, 493-495 (1995).
18 Choi, J.H., Ryu, Y.W., Park, Y.C., Seo, J.H., “Synergistic effects of chromosomal ispB deletion and dxs overexpression on coenzyme Q10 production in recombinant Escherichia coli expressing Agrobacterium tumefaciens dps gene”, J. Biotechnol., 144, 64-69 (2009).
19 Ye, J., Ma, L., Wu, H., Liu, X., Zhang, H., “Effect of overexpressing ubiCA genes responsible for ubiquinone biosynthesis on ubiquinone production in Escherichia coli”, J. East China Univ. Sci. Technol. (Natural Science Edition), 32, 269-273 (2006).
20 Cluis, C.P., Burja, A.M., Martin, V.J.J., “Current prospects for the production of coenzyme Q10in microbes”, Trends Biotechnol., 25, 514-521 (2007).
21 Huang, M.T., Wang, Y., Liu, J.Z., Mao, Z.W., “Multiple strategies for metabolic engineering of Escherichia coli for efficient production of coenzyme Q10”, Chin. J. Chem. Eng., 19, 316-326 (2011).
22 Zhong, W.H., Fang, J.J., Liu, H.G., Wang, X., “Enhanced production of CoQ10by newly isolated Sphingomonas sp. ZUTEO3 with a coupled fermentation-extraction process”, J. Ind. Microbiol. Biotechnol., 36, 687-693 (2009).
23 Noack, D., Roth, M., Geuther, R., Muller, G., Undisz, K., Hoffmeier, C., Gaspar, S., “Maintenance and genetic stability of vector plasmids pBR322 and pBR325 in Escherichia coli K12 strains grown in a chemostat”, Mol. Gen. Genet., 184, 121-124 (1981).
24 Bentley, W.E., Mirjalili, N., Andersen, D.C., Davis, R.H., Kompala, D.S., “Plasmid-encoded protein: the principal factor in the metabolic burden associated with recombinant bacteria”, Biotechnol. Bioeng., 35, 668-681 (1990).
25 O’Connor, M., Peifer, M., Bender, W., “Construction of large DNA segments in Escherichia coli”, Science, 244, 1307-1312 (1989).
26 Chiang, C.J., Chen, P.T., Chao, Y.P., “Replicon-free and markerless methods for genomic insertion of DNAs in phage attachment sites and controlled expression of chromosomal genes in Escherichia coli”, Biotechnol. Bioeng., 101, 985-995 (2008).
27 Tyo, K.E.J., Ajikumar, P.K., Stephanopoulos, G., “Stabilized gene duplication enables long-term selection-free heterologous pathway expression”, Nat. Biotechnol., 27, 760-765 (2009).
28 Datsenko, K.A., Wanner, B.L., “One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products”, Proc. Natl. Acad. Sci. USA, 97, 6640-6645 (2000).
29 Haldimann, A., Wanner, B.L., “Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structurefunction studies of bacteria”, J. Bacteriol., 183, 6384-6393 (2001).
30 Sharan, S.K., Thomason, L.C., Kuznetsov, S.G., Court, D.L.,“Recombineering: a homologous recombination-based method of genetic engineering”, Nat. Protoc., 4, 206-223 (2009).
31 Boyd, D., Weiss, D.S., Chen, J.C., Beckwith, J., “Towards singlecopy gene expression systems making gene cloning physiologically relevant: Lambda Inch, a simple Escherichia coli plasmid-chromosome shuttle system”, J. Bacteriol., 182, 842-847 (2000).
32 Lynch, H.C., Yang, Y., “Degradation products of clavulanic acid promote clavulanic acid production in cultures of Streptomyces clavuligerus”, Enzyme Microb. Tchnol., 34, 48-54 (2004).
33 Kwon, O., Hudspeth, E.S., Meganathan, R., “Anaerobic biosynthesis of enterobactin in Escherichia coli: Regulation of entC gene expression and evidence against its involvement in menaquinone (Vitamin K2) biosynthesis”, J. Bacteriol., 178, 3252-3259 (1996).
34 Buss, K., Müller, R., Dahm, C., Gaitatzis, N., Skrzypczak-Pietraszek, E., Lohmann, S., Gassen, M., Leistner, E., “Clustering of isochorismate synthase genes menF and entC and channeling of isochorismate in Escherichia coli”, Biochim. Biophys. Acta, 1522, 151-157 (2001).
35 Choi, J.H., Seo, Y.W., Seo, J.H., “Biotechnological production and applications of coenzyme Q10”, Appl. Microbiol. Biotechnol., 68, 9-15 (2005).
36 Jeya, M., Moon, H.J., Lee, J.L., Kim, I.W., Lee, J.K., “Current state of coenzyme Q10production and its applications”, Appl. Microbiol. Biotechnol., 85, 1653-1663 (2010).
37 Kien, N.B., Kong, I.S., Lee, M.G., Kim, J.K., “Coenzyme Q10production in a 150-L reactor by a mutant strain of Rhodobacter sphaeroides”, J. Ind. Microbiol. Biotechnol., 37, 521-529 (2010).
38 Goh, S., Good, L., “Plasmid selection in Escherichia coli using an endogenous essential gene marker”, BMC Biotechnol., 8, 61 (2008).
2012-12-10, accepted 2013-05-10.
* Supported by the National Natural Science Foundation of China (30970089, 20876181, 21276289) and the Natural Science Foundation of Guangdong Province (9351027501000003, S2011010001396).
** To whom correspondence should be addressed. E-mail: lssljz@mail.sysu.edu.cn
 Chinese Journal of Chemical Engineering2014年5期
Chinese Journal of Chemical Engineering2014年5期
- Chinese Journal of Chemical Engineering的其它文章
- Soft Sensor Model Derived from Wiener Model Structure: Modeling and Identification*
- Kinetics of Forward Extraction of Boric Acid from Salt Lake Brine by 2-Ethyl-1,3-hexanediol in Toluene Using Single Drop Technique*
- Influence of Solvent on Reaction Path to Synthesis of Methyl N-Phenyl Carbamate from Aniline, CO2and Methanol*
- Effect of Adsorbent Diameter on the Performance of Adsorption Refrigeration*
- High-Thermal Conductive Coating Used on Metal Heat Exchanger*
- A Facile Route for Synthesis of LiFePO4/C Cathode Material with Nano-sized Primary Particles*