
Temporal changes in the spinal cord transcriptome after peripheral nerve injury
2020-01-02 01:17:44JianWengDongDongLiBaoGuoJiangXiaoFengYin
中國神經(jīng)再生研究(英文版) 2020年7期
Jian Weng , Dong-Dong Li , Bao-Guo Jiang , Xiao-Feng Yin
1 Department of Orthopedics and Trauma, Peking University People's Hospital, Beijing, China
2 Department of Bone & Joint Surgery, Peking University Shenzhen Hospital, Shenzhen, Guangdong Province, China
3 Department of Surgery, the 517th Hospital of the People's Liberation Army, Xinzhou, Shanxi Province, China
Abstract Peripheral nerve injury may trigger changes in mRNA levels in the spinal cord. Finding key mRNAs is important for improving repair after nerve injury. This study aimed to investigate changes in mRNAs in the spinal cord following sciatic nerve injury by transcriptomic analysis.The left sciatic nerve denervation model was established in C57BL/6 mice. The left L4-6 spinal cord segment was obtained at 0, 1, 2, 4 and 8 weeks after severing the sciatic nerve. mRNA expression profiles were generated by RNA sequencing. The sequencing results of spinal cord mRNA at 1, 2, 4, and 8 weeks after severing the sciatic nerve were compared with those at 0 weeks by bioinformatic analysis. We identified 1915 differentially expressed mRNAs in the spinal cord, of which 4, 1909, and 2 were differentially expressed at 1, 4, and 8 weeks after sciatic nerve injury, respectively. Sequencing results indicated that the number of differentially expressed mRNAs in the spinal cord was highest at 4 weeks after sciatic nerve injury. These mRNAs were associated with the cellular response to lipid, ATP metabolism, energy coupled proton transmembrane transport, nuclear transcription factor complex, vacuolar proton-transporting V-type ATPase complex,inner mitochondrial membrane protein complex, tau protein binding, NADH dehydrogenase activity and hydrogen ion transmembrane transporter activity. Of these mRNAs, Sgk1, Neurturin and Gpnmb took part in cell growth and development. Pathway analysis showed that these mRNAs were mainly involved in aldosterone-regulated sodium reabsorption, oxidative phosphorylation and collecting duct acid secretion. Functional assessment indicated that these mRNAs were associated with inflammation and cell morphology development. Our findings show that the number and type of spinal cord mRNAs involved in changes at different time points after peripheral nerve injury were different. The number of differentially expressed mRNAs in the spinal cord was highest at 4 weeks after sciatic nerve injury. These results provide reference data for finding new targets for the treatment of peripheral nerve injury, and for further gene therapy studies of peripheral nerve injury and repair. The study procedures were approved by the Ethics Committee of the Peking University People's Hospital(approval No. 2017PHC004) on March 5, 2017.
Key Words: deep sequencing; expression profile; gene therapy; mRNAs; nerve regeneration; peripheral nerve injury; RNA sequencing; sciatic nerve injury; spinal cord; transcriptome
Introduction
Peripheral nerve injury is a common disease which may result in lifelong disability and physical dysfunction (Biazar et al., 2010; Weng et al., 2018). In the past few decades, the emergence of stem cells, biological materials, and nerve growth factors has greatly improved the repair of nerve damage, but this still does not meet clinical needs (Deumens et al., 2010; Sarker et al., 2018; Rao et al., 2019; Wu et al., 2019).The repair of peripheral nerve injury with satisfying outcomes is a major challenge in clinical practice. Recently, gene therapy has become an effective way to treat some diseases(Leers, 2019; Zhou et al., 2019). Exploring changes in mRNA levels in the spinal cord after nerve injury may be important for the development of novel treatments for peripheral nerve injury. The pathogenesis of peripheral nerve injury is associated with changes in RNAs, ion channels, receptors, and other intracellular proteins in neurons and supporting nerve cells (Akita et al., 2016; Uta et al., 2019). Nerve injury may trigger the transport of local signals from the peripheral injury site back to the cell body along axons (Abe and Cavalli,2008; Pathak et al., 2016). The subsequent regeneration of peripheral nerves has been directly attributed to intra-axonal translation of mRNAs. Moreover, these localized, quiescent mRNAs convert to translation mode following nerve injury(Mietto et al., 2015). Furthermore, the axonal mRNAs are translated to corresponding proteins that provide retrograde signals to the cell body (Perry and Fainzilber, 2014). However, it is still uncertain how this local translation is activated following peripheral nerve injury. The transcriptome is a crucial concept in the post-genomic era and has definite advantages for the study of peripheral nerve injury (Phay et al., 2015). Most cells share the same set of genes while their transcription is spatially and temporally specific. This is known as the dynamic evolution of mRNAs and it can be used for further investigations of the mechanisms underlying peripheral nerve injury to find novel means to treat nerve injury (Jiang et al., 2015).
Next-generation RNA sequencing (RNA-seq) is acknowledged as a highly sensitive method for the analysis of differentially expressed RNAs (Sirbu et al., 2012; Sun et al.,2017). Different from gene microarray assays, RNA-seq is not limited to a subset of known genes. RNA-seq can also analyze the entire genome of organs and tissues to provide information about previously unannotated genes as well as their possible functions (Hurd and Nelson, 2009; Zou et al.,2019). In addition, RNA-seq can detect smaller RNAs by not selecting for the poly-A tail (Guo et al., 2015). Although some studies have reported transcriptome changes in the stumps of lesioned sciatic nerve and dorsal root ganglia at several time points after injury (Yu et al., 2011), the temporal alteration of specific mRNAs directly responsible for nerve regeneration after injury remains unclear. The transcriptome and mRNA profile in the proximal nerve need to be further investigated.
In this study, transcriptome analysis of the spinal cord was performed using RNA-seq after sciatic nerve injury.The differentially expressed mRNAs after peripheral nerve injury were explored, the temporal change of mRNAs during peripheral nerve injury was investigated and the differentially expressed mRNAs were subjected to bioinformatic analyses to identify mRNAs related to post-injury nerve regeneration.
Materials and Methods
Animals
Fifteen healthy female wild-type C57BL/6J mice weighing 20-25 g and aged 6-8 weeks were used in this study [animals were bred in the Laboratory Animal Center of Peking University, animal use license No. SYXK (Jing) 2016-0009].The study procedures were approved by the Ethics Committee of the Peking University People's Hospital (approval No.2017PHC004) on March 5, 2017. This study was carried out in accordance with the principles of the Basel Declaration and recommendations of Chinese guidelines for the care and use of laboratory animals (China National Standardization Management Committee, No. GB/T 35823-2018, 2018). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). The mice were allowed free access to standard chow and water with a 12-hour light-dark cycle and standardized housing conditions before and after operation. Fifteen female C57BL/6 mice were randomly divided into five groups (0-, 1-,2-, 4-, 8-week groups, 0 weeks was the control group,n= 3 for each group).
Animal models and sample collection
The mice of all groups were fully anesthetized by isoflurane(RWD Life Science Co., Shenzhen, China) inhalation. A 0.5 cm incision was then made over the left hind limb sciatic nerve. The sciatic nerve was gently separated and exposed and cut at 1 cm from the branch of the tibial nerve and the common peroneal nerve, followed by closure of the incisions with 4-0 Ethilon sutures. All procedures were finished under aseptic conditions. To assess whether the model was successful, all mice were euthanized at 0 (control group), 1, 2, 4 and 8 weeks post-injury to ensure that the sciatic nerve was still completely disconnected. The L4-6 spinal cord on the injured side was collected and processed for further use.
RNA extraction and sequencing library construction
Fifteen spinal cord samples were collected at 0, 1, 2, 4 and 8 weeks (n= 3 at each time point) after injury. Total RNA was isolated using the RNeasy Mini Kit (Qiagen 217004, Dusseldorf, German) according to the manufacturer's instructions and the quality of the purified RNA evaluated with a Bio-Analyzer 2100 (Agilent Technology, CA, USA). The quantity of purified RNA was assessed using a NanoPhotometer spectrophotometer (IMPLEN, Westlake Village, CA, USA)at 260 nm. All RNA samples were stored at -80°C, and then sequenced on the Illumina platform by Novogene (Beijing,China). Sequencing libraries were prepared using 3 μg RNA per sample. Sequencing libraries were made using an NEBNext?UltraTMRNA Library Prep Kit for Illumina?(NEB,Ipswich, MA, USA) according to the manufacturer's instructions. Then, index codes were used to attribute sequences to each sample. First, poly-T oligo-attached magnetic beads were utilized to purify mRNA from total RNA. The divalent cations in NEBNext First Strand Synthesis Reaction Buffer(5×) were used for fragmentation under raising temperature.First-strand cDNA was synthesized using a random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-) and second-strand cDNA was subsequently made using DNA Polymerase I and RNase H. Overhangs were made blunt with polymerase/exonuclease. After the 3′ ends of DNA fragments were adenylated, NEBNext Adaptor with hairpin loop structure was ligated for hybridization. The library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, MA, USA). cDNA fragments of 150-200 bp were treated with 3 μL of USER Enzyme (NEB). The cDNA was ligated at 37°C for 15 minutes and then at 95°C for 5 minutes. Quantitative polymerase chain reaction (qPCR)was subsequently performed with Phusion High-Fidelity DNA polymerase, Universal qPCR primers and Index (X)Primer. Finally, the Agilent Bioanalyzer 2100 (Agilent Technology, CA, USA) system was used to evaluate the library quality and to purify the qPCR products.
Clustering and sequencing
Index-coded sample clustering was performed with TruSeq PE Cluster Kit v3-cBot-HS (Illumina, Santiago, CA, USA)following the instructions on the cBot Cluster Generation System. The libraries were sequenced to generate 125 bp/150 bp paired-end reads. For species with sophisticated gene annotation information (e.g., mice), reads could be directly mapped to the transcriptome. DNAStar Lasergene software(V2.5.1b; Madison, WI, USA) was used for comparison analysis of RNA-seq reads data. DNAStar used the Maximal Mappable Prefix search method to accurately position junction reads. HTSeq v0.6.0 was used to analyze differential gene expression of each sample. FPKM (expected number of fragments per kilobase of transcript sequence per million base pairs sequenced) is the number of fragments per kilobase of exon model per million mapped fragments, which takes the effect on sequencing depth and gene length on the fragment count into consideration, and is the most frequently used gene expression estimation method.
GO and KEGG enrichment analysis of differentially expressed mRNAs
Cluster Profiler R package was used to perform Gene Ontology (GO) enrichment analysis of differentially expressed genes, to revise the gene length bias. The enrichment of differentially expressed genes was considered statistically significant with a correctedP-value less than 0.05. Kyoko Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg) is a database for the identification of high-level functions of biological systems following genome sequencing or other high-throughput experimental technologies at the molecular level. The cluster Profiler R package was used to test the differentially expressed genes with significant enrichment in KEGG pathways.
Validation of expression for some differential mRNAs by qPCR
The remaining samples collected at 4 weeks were processed for validation by qPCR. The synthesis of first-strand cDNA was performed using a reverse transcription kit (Vazyme,Nanjing, China) according to the manufacturer's instructions. IQ SYBR green SuperMix reagent (Bio-Rad, Hercules,CA, USA) was chosen for amplification of target genes.Agarose gel electrophoresis and melting-curve analysis were employed to detect the specificity of qPCR. The comparative Ct method was used to quantify gene expression, and the relative quantification was calculated as 2-ΔΔCtwithGAPDHas an internal control. The primers used for qPCR are shown in Table 1.

Table 1 Primer sequences of GAPDH, Sgk1, Neurturin and Gpnmb genes
Statistical analysis
All data are expressed as the mean ± SEM. Statistical analysis was performed with SPSS version 19.0 software (IBM, Armonk, IL, USA). mRNA levels at different time points after injury were assessed using Student'st-test. The qPCR results were tested with Student'st-test. APvalue less than 0.05 was considered statistically significant.
Results
Number of sequence reads
After checking the RNA-seq data quality by determining the ratio of clean data (Figure 1A), the percentage of clean reads in the raw reads of each sample was more than 94%, indicating that the data were high-quality and able to be used for subsequent analysis. The distribution of reads in regions of the genome was also performed (Figure 1B). Different colors represent different genome regions, including exonic,intronic and intergenic regions.
Differentially expressed mRNAs after sciatic nerve injury
Differentially expressed mRNAs are independently displayed with a volcano plot, Venn diagram and clustering map (Figure 2). Compared with 0 week, the top ten differentially expressed mRNAs at 4 weeks and all differentially expressed mRNAs at 1 and 8 weeks after sciatic nerve injury are shown in Table 2. Differentially expressed mRNAs were not iden-tified at 2 weeks. We identified 1915 differentially expressed mRNAs after sciatic nerve injury compared with the control group (up-regulation: 2 at 1 week, 897 at 4 weeks, and 2 at 8 weeks; down-regulation: 2 at 1 week, 1012 at 4 weeks, and 0 at 8 weeks).

Table 2 Differentially expressed mRNAs in the spinal cord after sciatic nerve injury
Validation of expression of some differentially expressed mRNAs by qPCR
The expression of some differentially expressed mRNAs(Sgk1,NeurturinandGpnmb) at 1, 4 and 8 weeks after injury was validated by qPCR (Figure 3). The expression of these mRNAs was consistent with that determined by sequencing.
Functional prediction of differentially expressed mRNAs
To estimate the functions of mRNAs in the injured nerve,GO and KEGG pathway analysis were employed to analyze the threshold of fold change at > 0.95. GO analysis showed the most significantly enriched biological processes at 1 week were associated with cellular response to lipid, cellular response to organonitrogen compound, cellular response to nitrogen compound, cell growth and negative regulation of microtubule polymerization; at 4 weeks, ATP metabolic process, purine ribonucleoside triphosphate metabolic process, ribonucleoside triphosphate metabolic process, purine nucleoside triphosphate metabolic process and purine nucleoside metabolic process; and at 8 weeks, energy coupled proton transmembrane transport, ATP hydrolysis coupled proton transport, hydrogen ion transmembrane transport,proton transport and hydrogen transport. Of note, the most enriched cellular components at 1 week were associated with nuclear transcription factor complex; at 4 weeks, inner mitochondrial membrane protein complex, mitochondrial protein complex, mitochondrial inner membrane, organelle inner membrane and mitochondrial membrane part; and at 8 weeks, proton-transporting V-type ATPase complex,vacuolar proton-transporting V-type ATPase complex, proton-transporting domain, proton-transporting two-sector ATPase complex, proton-transporting two-sector ATPase complex and early endosome. The most significantly enriched molecular functions at 1 week were associated with protein kinase A catalytic subunit binding, tau protein binding, pre-mRNA binding, bHLH transcription factor binding and protein kinase A binding; at 4 weeks, NADH dehydrogenase activity, hydrogen ion transmembrane transporter activity, NADH dehydrogenase (ubiquinone) activity, NADH dehydrogenase (quinone) activity and oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor; and at 8 weeks, hydrogen ion transmembrane transporter activity, integrin binding, heparin binding, cell adhesion molecule binding and glycosaminoglycan binding (Figure 4). An enriched scatter diagram was used to demonstrate the results of Q-value, the Rich factor, KEGG enrichment analysis and number of genes reflected the degree of KEGG enrichment. According to the KEGG analysis results, the most significantly involved pathways in peripheral nerve injury at 1 week were aldosterone-regulated sodium reabsorption; at 4 weeks, metabolic pathways, Huntington's disease, oxidative phosphorylation, Parkinson's disease and Alzheimer's disease; and at 8 weeks, collecting duct acid secretion, rheumatoid arthritis, lysosome, oxidative phosphorylation and phagosome (Figure 5).
Scatterplots showed that the most significantly involved pathways in peripheral nerve injury at 4 weeks were metabolic pathways. The color of the dot indicates the size of theP-value. The size of the dot represents the number of genes.Padj indicates correctedP-value. The smaller the Padj value,the higher the correlation. “Count” represents the number of differentially expressed genes.
Discussion
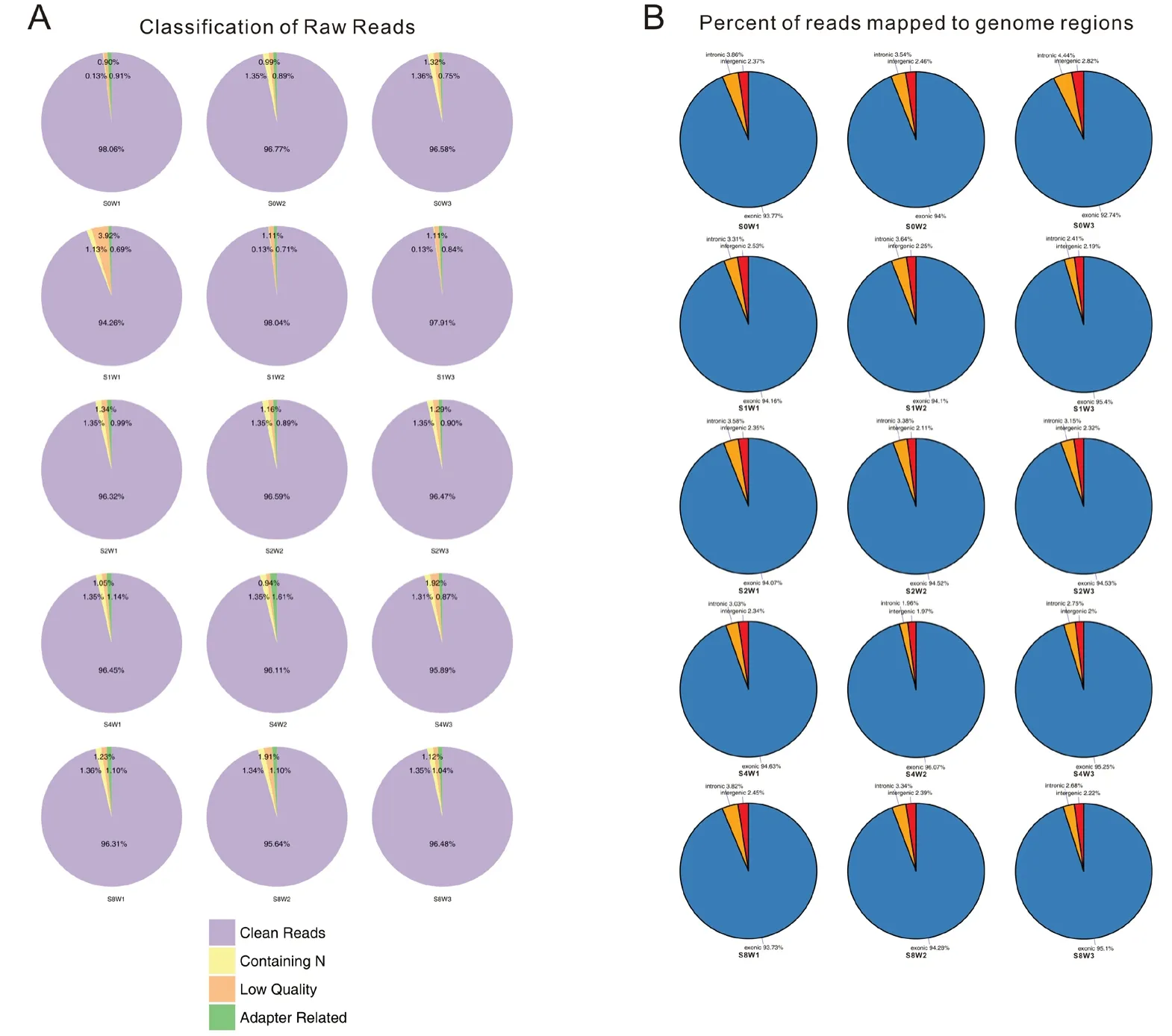
Figure 1 General information of the numbers and genomic localization of the sequence reads.(A) Percentage of clean reads in raw reads of each sample. Raw reads consist of clean reads, low quality reads, contaminating reads and adapter related reads.The percentage of clean reads in each sample was more than 94%and that of low-quality reads was less than 4%, indicating high quality data. (B) Percentage of reads mapped to genome regions. More than 93% of raw reads mapped to exon regions.
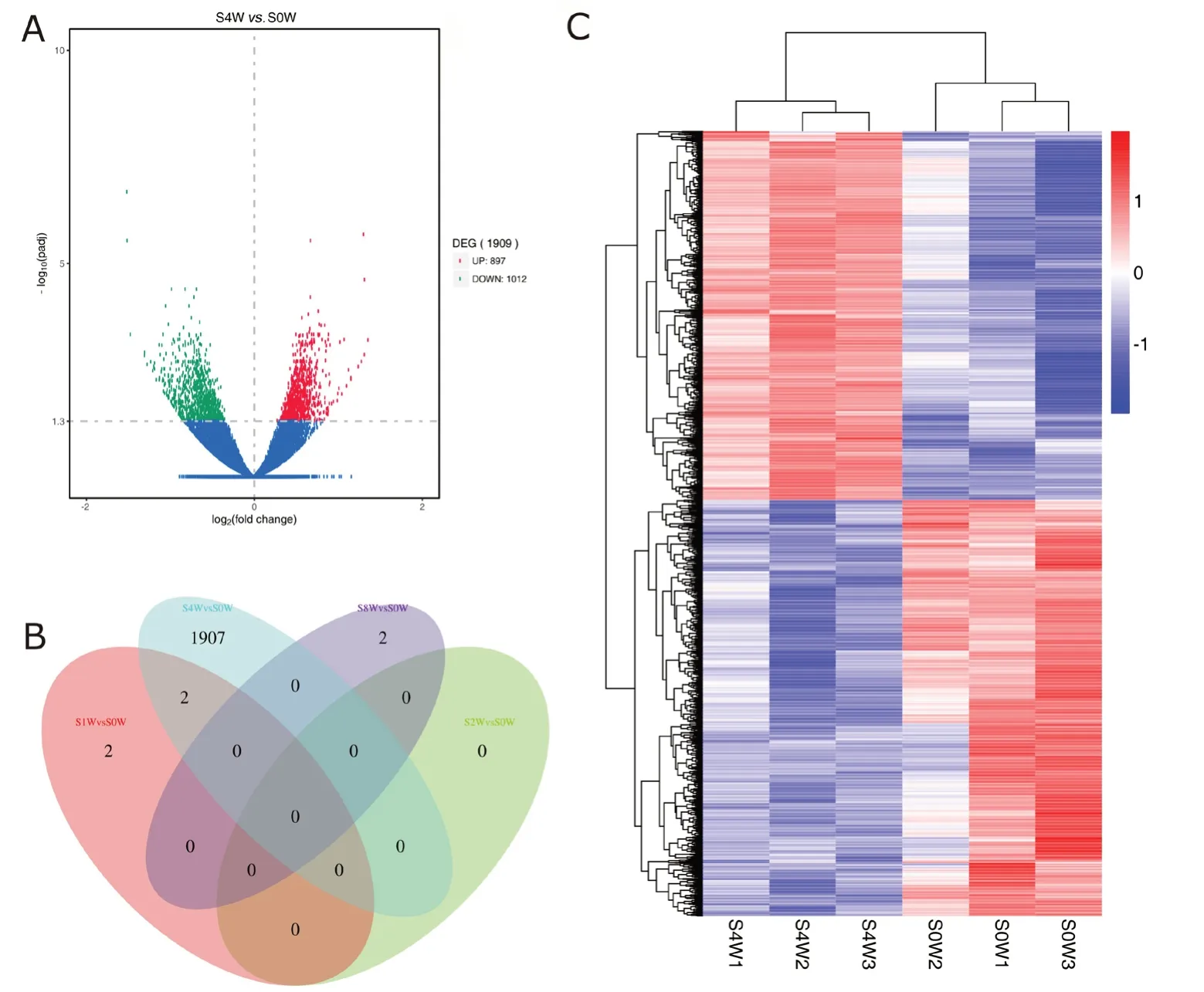
Figure 2 Differentially expressed mRNAs in the spinal cord at different time points after sciatic nerve injury.(A) Volcano plot shows the up-regulated and down-regulated mRNAs in the sciatic nerve injury group compared with control group at 4 weeks (red:up-regulation; green: down-regulation; blue: no significant change; DEG: differentially expressed gene). (B) Venn diagram indicates the number of overlapping differentially expressed mRNAs at 0, 1, 2, and 4 weeks. (C) Heat map of mRNAs displays the hierarchical clustering of differentially expressed mRNAs at 4 weeks (red: up-regulation; blue: down-regulation).
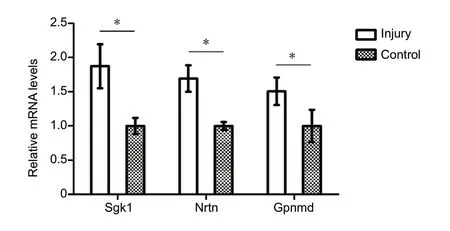
Figure 3 Validation of differentially expressed mRNAs by quantitative polymerase chain reaction at 4 weeks.The mRNA levels of serum/glucocorticoid regulated kinase 1 (Sgk1),neurturin (Nrtn) and glycoprotein (transmembrane) nmb (Gpnmb)were significantly up-regulated in the spinal cord after sciatic nerve injury. The quantitative polymerase chain reaction results were tested with Student's t-test. Data are presented as the mean ± SEM of three independent experiments (n = 3). *P < 0.05.
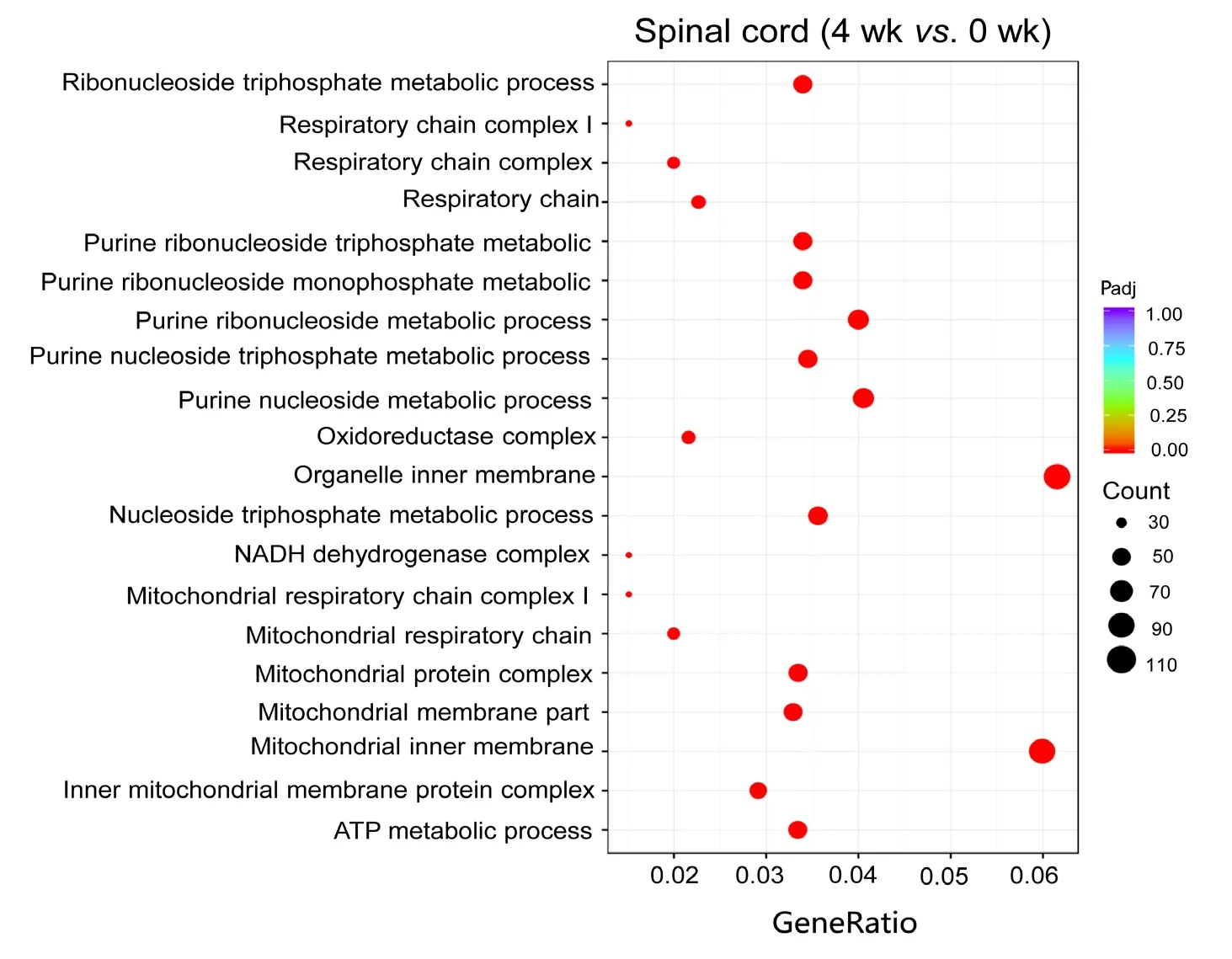
Figure 4 Gene Ontology analysis of differentially expressed mRNAs at 4 weeks.The node is used to represent the enrichment by color depth and circle size. Scatterplots displaying the most significantly enriched Gene Ontology terms are organelle inner membrane at 4 weeks. The color of the dot indicates the size of the Padj value. The size of the dot represents the number of genes. Padj indicates corrected P-value. The smaller the Padj value,the higher the correlation. “Count”represents the number of differentially expressed genes.
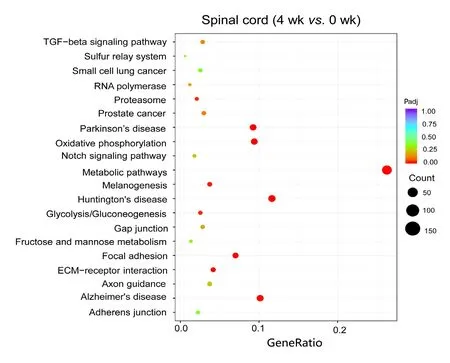
Figure 5 Kyoko Encyclopedia of Genes and Genomes analysis of differentially expressed mRNAs at 4 weeks.
Peripheral nerve injury is frequently seen in clinical settings,and incomplete neuroregeneration may lead to suboptimal recovery, including loss of limb movement and sensory dysfunction (Pannell et al., 2017; Lu et al., 2018; Sahar et al.,2019; Sturma et al., 2019). In recent decades, molecular biological studies have revealed some mechanisms of peripheral nerve regeneration following injury (Chen et al., 2019;Guo et al., 2019; Pina et al., 2019). This study investigated the RNA-seq profile in the spinal cord after injury. RNA-seq employs next generation high-throughput sequencing for the analysis of mRNA profiles (Metzker, 2010; Bahassi el and Stambrook, 2014), which can identify novel transcripts and disease-related genes. Our results revealed that the levels of some mRNAs changed significantly in the L4-6 spinal cord after injury, and the potential functions of differentially expressed mRNAs were predicted by GO and KEGG pathway analysis. These findings illustrate the dynamic change to mRNA profiles in peripheral nerve injury and regeneration.
After peripheral nerve injury, a characteristic pathological process known as Wallerian degeneration occurs in the injured nerve, in which there are complex interactions between Schwann cells and other cell types (such as macrophages, fibroblasts and mast cells which can secret inflammatory or anti-inflammatory cytokines (Rotshenker, 2011;Chen et al., 2015; DeFrancesco-Lisowitz et al., 2015; Zong et al., 2019). Wallerian degeneration is initiated immediately after peripheral nerve injury, in which ruptured axons and myelin are phagocytosed, followed by Schwann cell proliferation and formation of regenerated channels (Conforti et al., 2014; Jessen and Mirsky, 2016). The entire Wallerian degeneration process completes within approximately 4 weeks(Mietto et al., 2015). Peripheral nerve injury and Wallerian degeneration may trigger expression of mRNAs in the spinal cord. Some scholars refer to this phenomenon as spinal cord remodeling (Gu et al., 1998; Peixun et al., 2017). Recent studies have found that regulating the expression of some proteins in the spinal level can also promote the regeneration of peripheral nerves (Gallaher and Steward, 2018; Huang et al., 2018). However, limited by first generation sequencing technology, not all the genes in the spinal cord involved in peripheral nerve damage have been identified. This study used the most advanced sec-ond-generation sequencing technology to determine mRNAs changes in the spinal cord.
In this study, only four differentially expressed mRNAs were identified in the spinal cord in the first 2 weeks after sciatic nerve injury (4 after 1 week and 0 after 2 weeks),while 1909 appeared after 4 weeks, and then two were found after 8 weeks. Further analysis revealed that the dynamic temporal change of mRNA expression reflected alteration of some important biological processes in the L4-6 spinal cord during the 4-week Wallerian degeneration period. Several differentially expressed genes exert various effects on certain biological processes, such as inflammation, cell morphology development functions (such asSgk1,NeurturinandGpnmb)and nerve regeneration. GO analysis is a primary bioinformatics tool that can unify the representation of genes and gene products (Altshuler et al., 2008). GO terms and GO notes can predict gene function (Du et al., 2016).
The KEGG database can provide functional information for systematic analysis and is therefore widely used as a platform for enrichment analysis of gene functions (Camon et al., 2004). The functions of related genes and the pathways related to differentially expressed mRNAs in the L4-6 spinal cord after sciatic nerve injury were further analyzed using GO and KEGG. Our results revealed that the biological processes strongly associated with the pathogenesis of peripheral nerve injury were cell growth, mitochondrial synthesis,tricarboxylic acid cycling and neurological regeneration. In addition, the pathways significantly involved in the pathogenesis of peripheral nerve injury were aldosterone-regulated sodium reabsorption, oxidative phosphorylation and collecting duct acid secretion. KEGG and GO analysis showed the pre-dicted functions of differentially expressed mRNAs in peripheral nerve repair.
Nevertheless, this study has some limitations. First, the expression profiles of the biological replicates are divergent at 1, 2, and 8 weeks. All the samples were harvested by one experienced surgeon and the other variables were strictly controlled (age, sex, external appearances, animal genetic background, and feeding conditions). We speculate that the expression of mRNAs in the spinal cord is affected by the time of injury after peripheral nerve injury. Second, further experiments and study designs are not presented. We have been working on discovering new gene targets related to peripheral nerve injury repair and central nervous system remodeling. Our results have established a reliable information platform that can be used as the basis for further study.We hope that the results we present here may be useful for other teams who are interested in this field. We have also preparedin vitroandin vivoexperiments to validate some of the interesting findings of our high-throughput experiments.
High-throughput sequencing enables investigation of the dynamic temporal change of mRNA expression profiles in the L4-6 spinal cord after peripheral nerve injury. Furthermore, the differentially expressed mRNAs in the proximal spinal cord were explored and the results showed a local inflammatory response in Schwann cells or other related cells during post-injury nerve repair. The number and type of spinal cord mRNAs involved in changes at different time points after peripheral nerve injury were different. These findings provide targets for future treatment of peripheral nerve injury at different stages.
Author contributions:Study design: XFY and BGJ; experimental implementation: JW; data analysis: JW and DDL; paper writing: JW and DDL.All authors approved the final version of the paper.
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support:This research was supported by the National Natural Science Foundation of China, No. 81671215 (to XFY), No. 31571002 (to BGJ); the Natural Science Foundation of Beijing of China, No. 7192215 (to XFY). The funding sources had no role in study conception and design,data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement:The study procedures were approved by the Animals Ethics Committee of the Peking University People's Hospital, China (approval No. 2017PHC004) on March 5, 2017. This study was carried out in accordance with the principles of the Basel Declaration and recommendations of Chinese guidelines for the care and use of laboratory animals (China National Standardization ManagementCommittee, No. GB/T 35823-2018, 2018). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
- 中國神經(jīng)再生研究(英文版)的其它文章
- Diabetic retinopathy: a matter of retinal ganglion cell homeostasis
- Insights from human sleep research on neural mechanisms of Alzheimer's disease
- Protective effects of pharmacological therapies in animal models of multiple sclerosis: a review of studies 2014-2019
- EDITORIAL BOARD
- Recovery of injured corticoreticulospinal tract following cranioplasty in an ischemic stroke patient: a diffusion tensor tractography study
- Neurotoxic role of interleukin-17 in neural stem cell differentiation after intracerebral hemorrhage