
The Wolfiporia cocos Genome and Transcriptome Shed Light on the Formation of Its Edible and Medicinal Sclerotium
2020-07-05 07:36:22HongmeiLuoJunQianZhichaoXuWanjingLiuLeiXuYingLiJiangXuJianhongZhangXiaolanXuChangLiuLiuHeJianqinLiChaoSunFrancisMartinJingyuanSongShilinChen
Hongmei Luo, Jun Qian, Zhichao Xu, Wanjing Liu, Lei Xu, Ying Li Jiang Xu, Jianhong Zhang, Xiaolan Xu, Chang Liu, Liu He, Jianqin Li Chao Sun, Francis Martin*, Jingyuan Song,*,Shilin Chen,,*
1 Engineering Research Center of Chinese Medicine Resource, Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100193, China
2 College of Pharmacy, Hubei University of Chinese Medicine, Wuhan 430065, China
3 Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China
4 INRA, Universite′ de Lorraine, UMR 1136 Interactions Arbres/Microorganismes, 54280 Champenoux, France
5 Beijing Advanced Innovation Center for Tree Breeding by Molecular Design, Institute of Microbiology, Beijing Forestry University, Beijing 100083, China
KEYWORDS Wolfiporia cocos;Genome sequencing;Sclerotial formation;Fungal development;Polysaccharide and triterpenoid biosynthesis
Abstract Wolfiporia cocos (F.A.Wolf) has been praised as a food delicacy and medicine for centuries in China.Here,we present the genome and transcriptome of the Chinese strain CGMCC5.78 of W.cocos.High-confidence functional prediction was made for 9277 genes among the 10,908 total predicted gene models in the W. cocos genome. Up to 2838 differentially expressed genes (DEGs)were identified to be related to sclerotial development by comparing the transcriptomes of mycelial and sclerotial tissues.These DEGs are involved in mating processes,differentiation of fruiting body tissues, and metabolic pathways. A number of genes encoding enzymes and regulatory factors related to polysaccharide and triterpenoid production were strikingly regulated. A potential triterpenoid gene cluster including the signature lanosterol synthase(LSS)gene and its modified components were annotated. In addition, five nonribosomal peptide synthase (NRPS)-like gene clusters,eight polyketide synthase (PKS) gene clusters, and 15 terpene gene clusters were discovered in the genome. The differential expression of the velevt family proteins, transcription factors,carbohydrate-active enzymes, and signaling components indicated their essential roles in the regulation of fungal development and secondary metabolism in W. cocos. These genomic and transcriptomic resources will be valuable for further investigations of the molecular mechanisms controlling sclerotial formation and for its improved medicinal applications.
Introduction
Wolfiporia cocos(F.A.Wolf)Ryvarden&Gilb.,a member of the Polyporaceae family, is one of the medicinal mushrooms forming edible sclerotium. W. cocos is also a well-known brown rot species widely distributed around the world [1,2].As one of the Aphyllophorales fungi, W. cocos parasitizes the roots of conifers (e.g., Picea, Tsuga, and Pinus) and hardwood trees (e.g., Citrus, Eucalyptus, Quercus, and Fagus). The large sclerotium is formed close to the roots of the host[3,4]or on basswood that is inoculated and stored in caverns [3]. The large edible sclerotium of W. cocos has been referred to as‘‘Indian bread”or‘‘tuckahoe”in North America[3].W.cocos is widely used as nutraceuticals, cosmetics, tea supplements,and functional food [5,6]. Dried sclerotium, which is known as Fuling in traditional Chinese medicine(TCM),is also widely used as a crude drug in East Asia.
Fungal sclerotia are hard, asexual, and long-lived resting structures composed of aggregated vegetative hyph? [9]. The dry compact biomass of W. cocos sclerotium is over 80% of fibers, mainly composed of β-D-glucan-type nonstarch polysaccharides [7,8]. The developmental mechanisms leading to sclerotial formation have been investigated in Sclerotinia sclerotiorum, a model system for the study of sclerotial development and formation [9,10]. Generally, oxidative stress, low pH, and hyphal damage trigger sclerotial formation [9,11,12].In addition to environmental changes, primary metabolism,secondary messengers, and molecular components also play important roles in regulating sclerotial development [9]. The development of W.cocos sclerotium entails complex multistep processes related to peroxisomes, fatty acid desaturation, and degradation pathways [13].
The dried sclerotium of W. cocos is a source of many secondary metabolites, including polysaccharides and triterpenoids, which are major ingredients and bioactive compounds of pharmaceuticals[3,5,6,14].The polysaccharides isolated from polypore fungi with high molecular weight are derived from fungal cell walls [15]. These compounds exhibit anti-inflammatory effects [16], immunomodulatory properties[17,18], and anticancer activities [5]. A large number of(1-3)-β-D- and (1-6)-β-D-glucan-type polysaccharides and lanostane/secolanostane skeleton-derived triterpenoids have been isolated from W. cocos [3,19]. Although the genome of an American strain of W. cocos has been previously released by the Joint Genome Institute (JGI) [20], its features have not been discussed. Regulation of pathways involved in lignocellulose decomposition and triterpenoid synthesis has been investigated [21–23], but the molecular mechanisms involved in sclerotial development and biosynthesis of secondary metabolites remain largely unknown.
Herein,we report the analysis of the genome and transcriptome of a Chinese strain CGMCC5.78 of W. cocos. In addition, we compare this draft genome to the genome of the American strain [20] and to other related fungi (Table S1).Considering the specific feature of the edible and medicinal sclerotia of W. cocos, this comparative genome analysis will increase our knowledge about the molecular mechanisms involved in sclerotial formation and bioactive compound biosynthesis in W. cocos. Furthermore, these data will open up new avenues for the investigation of genetic breeding and commercial production of the edible and medicinal W. cocos sclerotia.
Results
Genomic features of W. cocos
We sequenced the genome of W.cocos(CGMCC5.78)by using HiSeq 2000 Illumina short-reads sequencing and a fosmid-tofosmid strategy. The size of the final genome assembly is estimated to 50.6 Mb(Table 1,Tables S2 and S3).We mapped the short reads against the genome assembly by SOAPaligner [24]to assess the sequencing depth. The percentage of reads with sequencing depths lower than 10× was less than 2% among the total genomic reads (Figure S1). We also sequenced five randomly selected fosmid clones to assess the accuracy of the genome assembly, and found that their sequences were nearly identical to the corresponding genome scaffolds (Table S4).Additionally, a large number of transposable elements (TEs)were found in the assembled genome, and the most abundant type of TEs was long terminal repeats (LTRs), accounting for~20%of the assembly(Table 2).Moreover,10,908 gene models were prediected(Table S5),of which 9277 genes(85%)were functionally annotated (Table S6). tRNAs, rRNAs, and snRNAs were also annotated, and 29 of the 184 tRNAs were identified to be pseudogenes (Table S7).
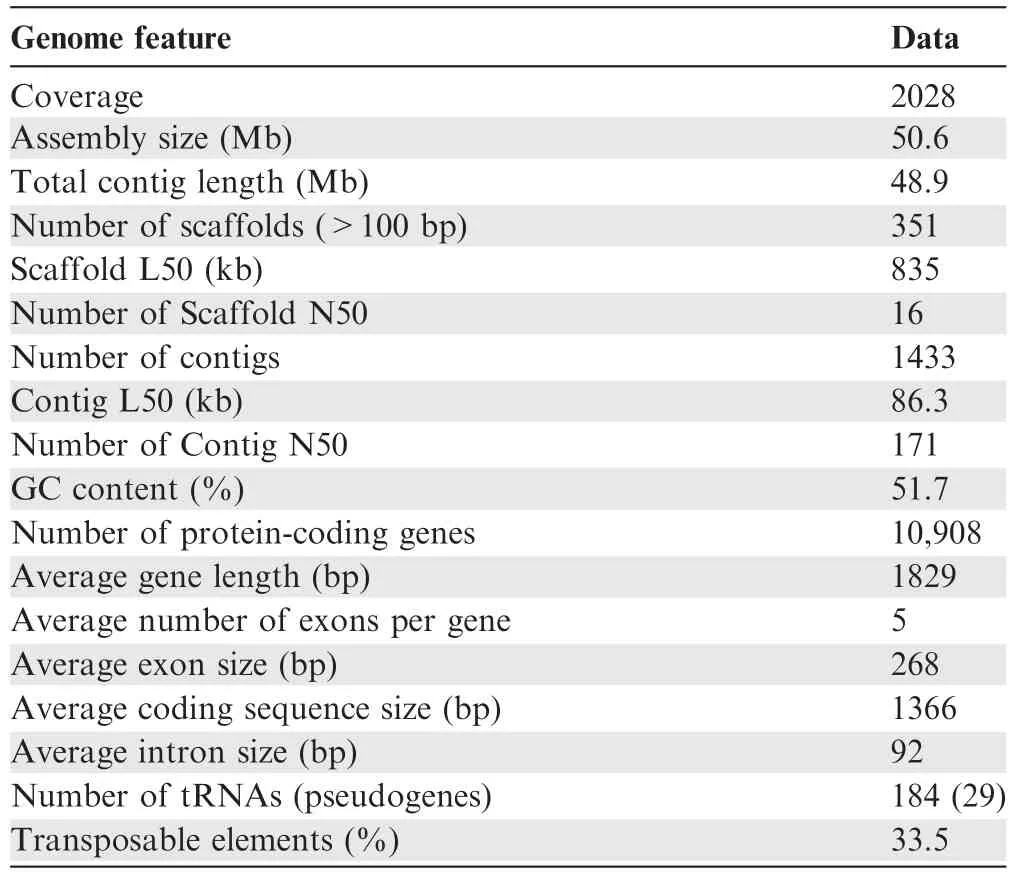
Table 1 Genome characteristics of W. cocos
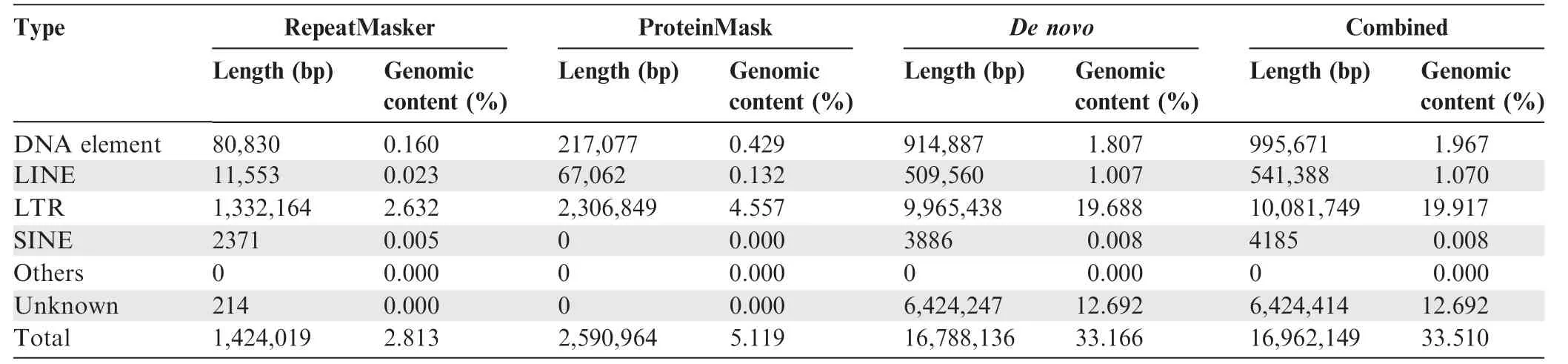
Table 2 Repeat content in the assembled W. cocos genome
The numbers and proportions of single-copy orthologs,multiple-copy orthologs, unique paralogs, other orthologs,and unclustered genes in the genomes of W. Cocos and other fungi were shown in Figure 1A and Table S8. 1440 speciesspecific genes and 234 unique gene families were identified in W. cocos by comparative genome analysis against other 14 fungi using OrthoMCL [25] (Table S9A). These speciesspecific genes are enriched in the Gene Ontology (GO) terms of ‘‘metabolic process”, ‘‘binding”, and ‘‘catalytic activity”(Table S9B)and in the Kyoto Encyclopedia of Genes and Genomes(KEGG)pathway of‘‘metabolism”(Table S9C).A total of 1868 orthologs existed in the genomes of W. cocos, Postia placenta, Phanerochaete chrysosporium, and Schizophyllum commune,while 377 genes were specific to W.coco(Figure 1B).
As expected, the W. cocos genomes of the Chinese strain CGMCC5.78 [hereafter referred to as W. cocos (IMPLAD)]and the American strain MD-104 SS10 [hereafter referred to as W. cocos (JGI)] [20] are highly similar with a high average similarity of 92% for the aligned regions (such as scaffold 1,Figure 2A and B).The number of predicted genes for W.cocos(IMPLAD) (10,908 genes) is slightly lower than that of W.cocos (JGI) (12,746 genes) [20], possibly due to the use of different gene prediction programs,but also the known heterozygosity of W.cocos(IMPLAD).The comparison of OrthoMCL gene families(Table S10)and whole genomes(Table S11)confirmed the genetic differences between these two W. cocos strains, as illustrated by the phylogenetic analysis (Figure 2C)and the alignment of the rDNA ITS2 sequences (Figure S2).The number of single-copy orthologs,multiple-copy orthologs,and unique paralogs between these two strains are nearly identical (Figure S3A). A total of 6575 shared orthologous groups were identified in the genomes of these two fungi(Figure S3B).A phylogenetic tree of the genomes of both W. cocos strains and the other sequenced fungi (P. placenta, P. chrysosporium,Laccaria bicolor, and S. commune) was constructed using single-copy orthologous genes (Figure S3C). The molecular clock for the first phase sites in each species was calculated with single-copy orthologous genes. The divergence time between the two W.cocos strains was estimated to be 19.3 million years ago (MYA) (Figure S3C). The difference in gene family expansion and contraction in these strains was also detected by CAFE [26], and the result showed that, of the 6891 gene families presented in the most recent common ancestor of the six fungal species, 154 gene families in W. cocos(IMPLAD) exhibited expansion, whereas 1387 genes families exhibited contraction; in comparison, 284 gene families in W.cocos (JGI) exhibited expansion, whereas 127 genes families exhibited contraction(Figure S3D).The observed genomic differences might be associated with the phenotypic and physiological differences between the Chinese and American W.cocos strains.

Figure 1 Genomic features of W. cocosA.The number of single-copy orthologs,multiple-copy orthologs,unique paralogs,other orthologs,and unclustered genes in Aspergillus nidulans, Coprinopsis cinerea, Cryptococcus gattii, Cryptococcus neoformans, Laccaria bicolor, Melampsora laricis-populina, Neurospora crassa, Phanerochaete chrysosporium, Postia placenta, Puccinia graminis, Saccharomyces cerevisiae, Schizophyllum commune, Serpula lacrymans,Ustilago maydis,and Wolfiporia cocos.B.The orthologs identified in W.cocos,P.placenta,P.chrysosporium,and S.commune by OrthoMCL after BLASTP (E value < 1E–5).
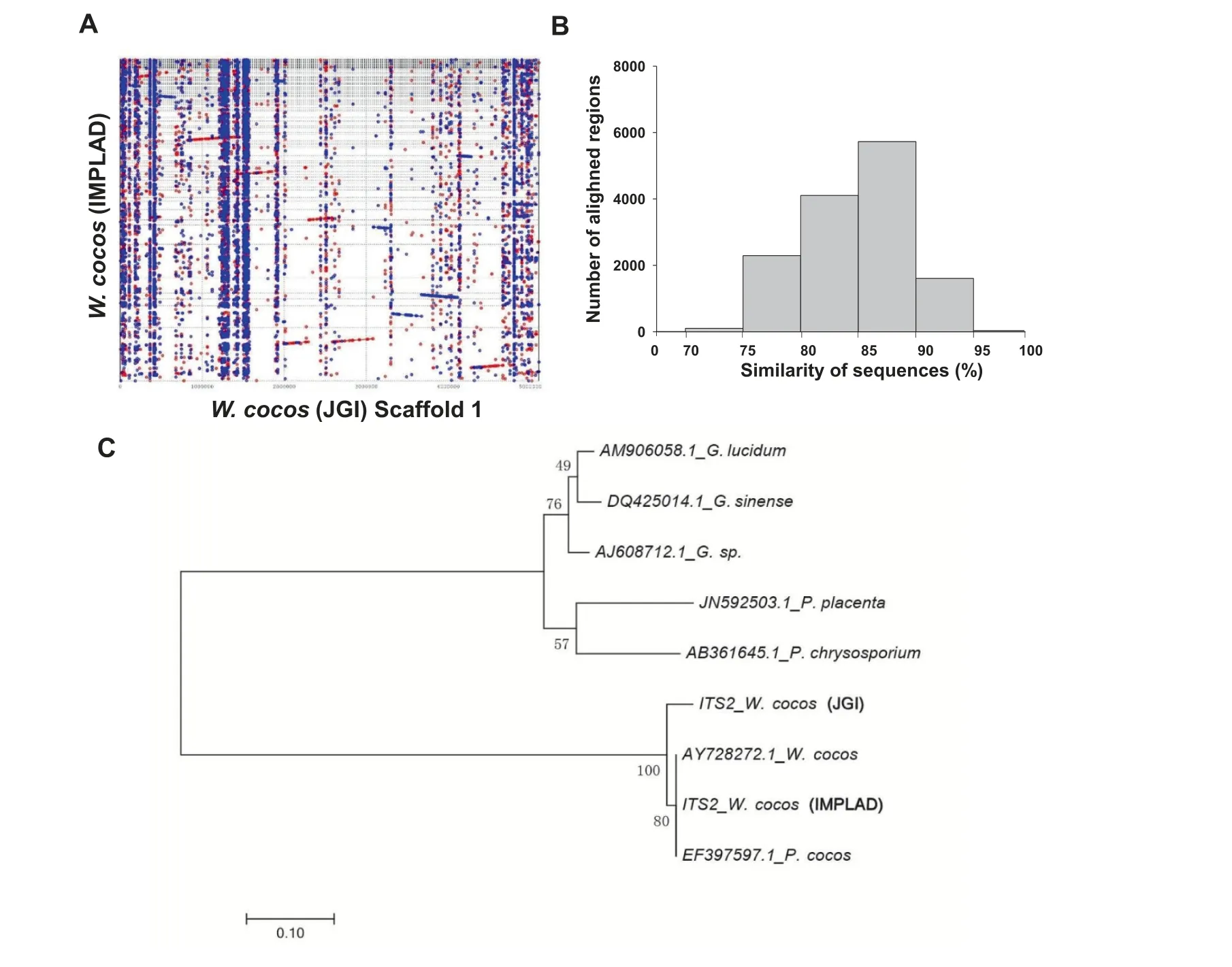
Figure 2 Genomic comparison of the Chinese and American strains of W. cocosA.Synteny dot plot showing the comparison of the whole genome of W.cocos(sequenced by IMPLAD in this study)to scaffold 1 of the American W. cocos genome (sequenced by JGI). B. Histogram showing similarity distribution of the 13,874 aligned regions. C.Evolutionary relationships of different W. cocos strains with other Polyporales species including Ganoderma sp., Ganoderma lucidum,Ganoderma sinense, P. chrysosporium, and P. placenta by ITS2 sequences. The sequences of AY728272.1 and EF397597.1 for ITS2 were downloaded from GenBank.The ITS2 sequences of the other two W.cocos strains were isolated from the W.cocos genomes sequenced by JGI and IMPLAD (CGMCC5.78), respectively. The phylogenetic tree was constructed using the neighbor-joining method. The percentages of replicated trees in which the associated taxa were clustered together in the bootstrap test(1000 replicates)are shown next to the branches. IMPLAD, Institute of Medicinal Plant Development; JGI, Joint Genome Institute.
Transcript profiling
To uncover the molecular mechanisms related to secondary metabolite biosynthesis and sclerotial development, we compared the gene expression profiles between the vegetative mycelium and the sclerotium of W. cocos (IMPLAD) (Figure 3A).Among the total RNA reads produced from the mycelium and sclerotium, 73%and 65%of the reads were mapped to the genome, respectively (Table S12). A total of 8548 genes were expressed in the vegetative mycelium (8395 genes) and sclerotium(8479 genes)(Table S13).A relative high sequencing coverage was presented in most of the genes in mycelium and sclerotium(Table S14).A total of 2838 genes showed differential expression in the sclerotium tissues compared to the mycelium tissues: 1877 were upregulated, whereas 961 were downregulated (Table S15). The GO and KEGG enrichment analyses showed that genes involved in ‘‘cell/cell part”, ‘‘catalytic activity”, ‘‘metabolic process” (Table S16), and ‘‘metabolic pathways” (Table S17) were prominent. To validate the RNA-seq transcript profiling, the relative expression levels of ten key genes involved in the mevalonate (MVA) pathway were measured by RT-qPCR. The relative expression of seven of these marker genes was consistent with the RNA-seq data(Table S18).
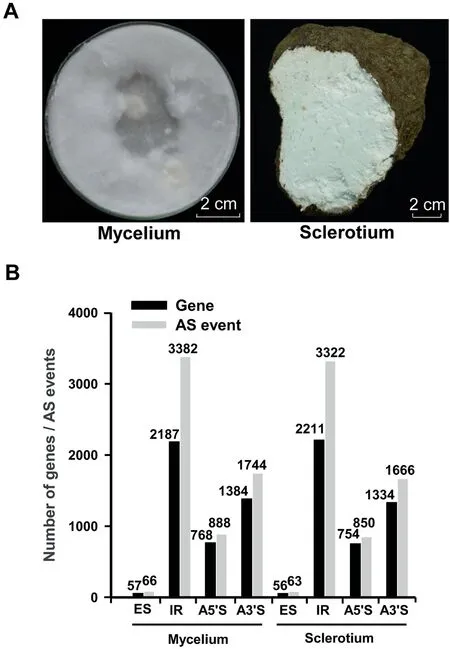
Figure 3 Transcriptomic analysis of W. cocosA. The 7-day-old mycelium and the one-year-old fresh sclerotium of W. cocos used for transcriptome analysis. B. The number of genes for AS events in the transcriptomes of W. cocos mycelium and sclerotium. AS, alternative splicing; ES, exon skipping; IR,intron retention; A5?S, alternative 5?splicing; A3?S, alternative 3? splicing.
Alternative splicing(AS)events are widespread in the transcriptome of W. cocos. Intron retention (IR) and exon skipping (ES) are the largest and least variable AS types in the transcriptomes of vegetative mycelium and sclerotium tissues(Figure 3B, Tables S19 and S20). In addition, a total of 3539 and 3809 novel transcript units (TUs) were predicted in the transcriptomes of the mycelium and sclerotium, respectively(Table S21).
Genes related to sclerotial development
Gene families of G-protein-coupled receptors (GPCRs), heterotrimeric G protein subunits (Gα subunit), and monomeric GTPase modules (RhoGEF family), known to be involved in sclerotial development in S.sclerotiorum[9,10],displayed gene expansion in Polyporaceae(e.g.,P.placenta,P.chrysosporium,Ganoderma lucidum, and W. cocos) compared to Sclerotiniaceae (e.g., S. sclerotiorum and Botrytis cinerea). In contrast,the histidine kinase gene family is narrowed in Polyporaceae(Table S22).The diversification of these gene families suggests that developmental pathways involved in sclerotial formation in W. cocos are likely different to pathogenic sclerotia. In the sclerotium of W. cocos, the expression of 21 signaling genes was upregulated (e.g., Gα subunit, RasGEF, RhoGEF, Rho-GAP, MAPK, and MAPKK), whereas the expression of 13 was downregulated (e.g., STE3, Gα subunit, RGS, and Rho-GEF) compared to that in the mycelium, suggesting that they play a key role in the developmental transition from mycelium to sclerotium.In contrast,orthologous genes involved in metabolic pathways were shared between W. cocos and sclerotialproducing fungi S. sclerotiorum and B. cinerea (Table S23),indicating that several metabolic processes have been coopted in sclerotia produced by taxonomically divergent species.
Genes related to the mating process
The mating process in Basidiomycota is coordinated by key homeodomain (HD) genes and pheromone recepoter genes[27]. A candidate HD1-HD2 gene pair (WCO006754.1-WCO006755.1) was annotated in W. cocos (IMPLAD) genome(Figure S4;Table S24).These two HD genes were located in scaffold 4, 73 kb away from a conserved mitochondrial intermediate peptidase gene(MIP;WCO006733.1)(Figure S4;Table S24), while a conserved fungal gene of unknown function (β-FG, WCO000491.1) was located in scaffold 1(Table S24). Interestingly, another HD-like gene HD3(WCO006732.1) was located near the MIP gene in scaffold 4.In contrast,a MIP gene and a β-FG gene were both located in scaffold 1 and no HD genes were identified in W. cocos(JGI) genome (Figure S4; Table S24). Furthermore, two STE3-like pheromone receptor genes (WCO006868.1 and WCO007080.1) existed in different scaffolds of W. cocos(IMPLAD) genome. Similarly, four STE3-like pheromone receptor genes were also located in different scaffolds of W.cocos (JGI) genome (Figure S4; Table S24).
Genes related to fruiting body formation
A white or slightly yellow structure,similar to a honeycomb,is the sexual reproductive organ of W. cocos (Figure S5). This fruiting body usually grows on the surface of wood, soil, or culture media and produces spores.The morphological characteristics of the fruiting body are the basis for the classification of W.cocos.According to a previous study on the formation of fruiting bodies in other fungi [10], the signalosome including sexual development subunits, transcription factors (TFs),mutanase, fatty acid oxygenase, and transcriptional activators might play a role in the development and formation of fruiting bodies.The homologous genes of these components have been identified in W. cocos genome (Table S25).
Genes involved in secondary metabolism
Genes related to polysaccharide biosynthesis and regulation
In W. Cocos, (1-3)-β-D and (1-6)-β-D glucans are the major components of the bioactive polysaccharides, and their concentrations impact the quality and value of W.cocos sclerotia.The biosynthesis of these glucans was extensively studied in yeast [28,29]. (1-3)-β glucans are synthesized from UDPglucose by the (1-3)-β glucan synthase (GS) complex(EC:2.4.1.34) [30], which is composed of a catalytic subunit(Fksp, encoded by FKS1 [31,32]) and a regulatory subunit(Rho1p) [33] in Saccharomyces cerevisiae. Genes involved in biosynthesis of (1-3)-β-D and (1-6)-β-D glucans are depicted in Figure 4A and B,together with a phosphoglucomutase gene(WCO006270.1) that is highly expressed in the sclerotium(Table S26).
Fksp (EC: 2. 4.1.34) uses UDP-glucose to produce a linear polysaccharide[28]. There are two glucan synthase genes,designated as FKS1 (WCO003877.1) and FKS2 (WCO000998.1),in the W. cocos (IMPLAD) genome; however, only FKS1 displayed differential expression during sclerotial development(Table S26). FKS1 was upregulated in sclerotium compared to mycelium, indicating its essential role in the regulation of polysaccharide biosynthesis and sclerotial development in W.cocos (Table S26). Phylogenetic analysis of FKS proteins suggests that this enzyme is highly conserved in fungi and, as expected,FKS from the Chinese and American strains showed the highest homology (Figure 4C; Table S27).
The genes responsible for the regulation of S.cerevisiae cell wall polysaccharide content have been characterized [28,29].Several of these genes and gene families were likely involved in the regulation of polysaccharide biosynthesis in W. cocos,includingRHO1multicopysuppressor(ROM1;WCO006350.1) and killer toxin resistant 6 (KRE6;WCO007777.1 and WCO009633.1) (Table S26). Moreover,these three genes were highly expressed in the sclerotium,which might be relevant to the higher polysaccharide content in sclerotium.
Genes related to triterpenoid biosynthesis
Pachymic acid(PA),a lanostane-type triterpenoid with a wide range of bioactivities,is synthesized via the mevalonate(MVA)pathway(Figure 5A).All genes involved in the MVA pathway were annoated in W. cocos genome, including several gene families [e.g., acetyl-CoA acetyltransferase (AACT) family,farnesyl diphosphate synthase (FPPS)family, and diphosphomevalonate decarboxylase (MVD) family], and single-copy genes [e.g., 3-hydroxy-3-methylglutaryl-coenzyme A reductase(HMGR), hydroxymethylglutaryl-CoA synthase A (HMGS),lanosterol synthase (LSS), and squalene monooxygenase(SE)] (Table S28). Upregulation of most of these genes was induced by methyl jasmonate (MeJA) for 2 h treatment and then downregulation occurred after 12 h treatment(Figure 5B);however, the expression of AACT1 (WCO010060.1), FPPS3(WCO002210.1),MVD1(WCO001214.1),MVD3(WCO006131.1), and MVD4 (WCO006124.1) were not detected in this assay(data not shown).In addition,transcriptome analysis showed that AACT1 (WCO010060.1), HMGR(WCO008156.3),HMGS(WCO006829.1),MVD2(WCO004278.1), and SE (WCO000689.1) were more highly expressed in mycelium than in sclerotium,implying the important function of these genes in triterpenoid biosynthesis in W.cocos (Table S28).
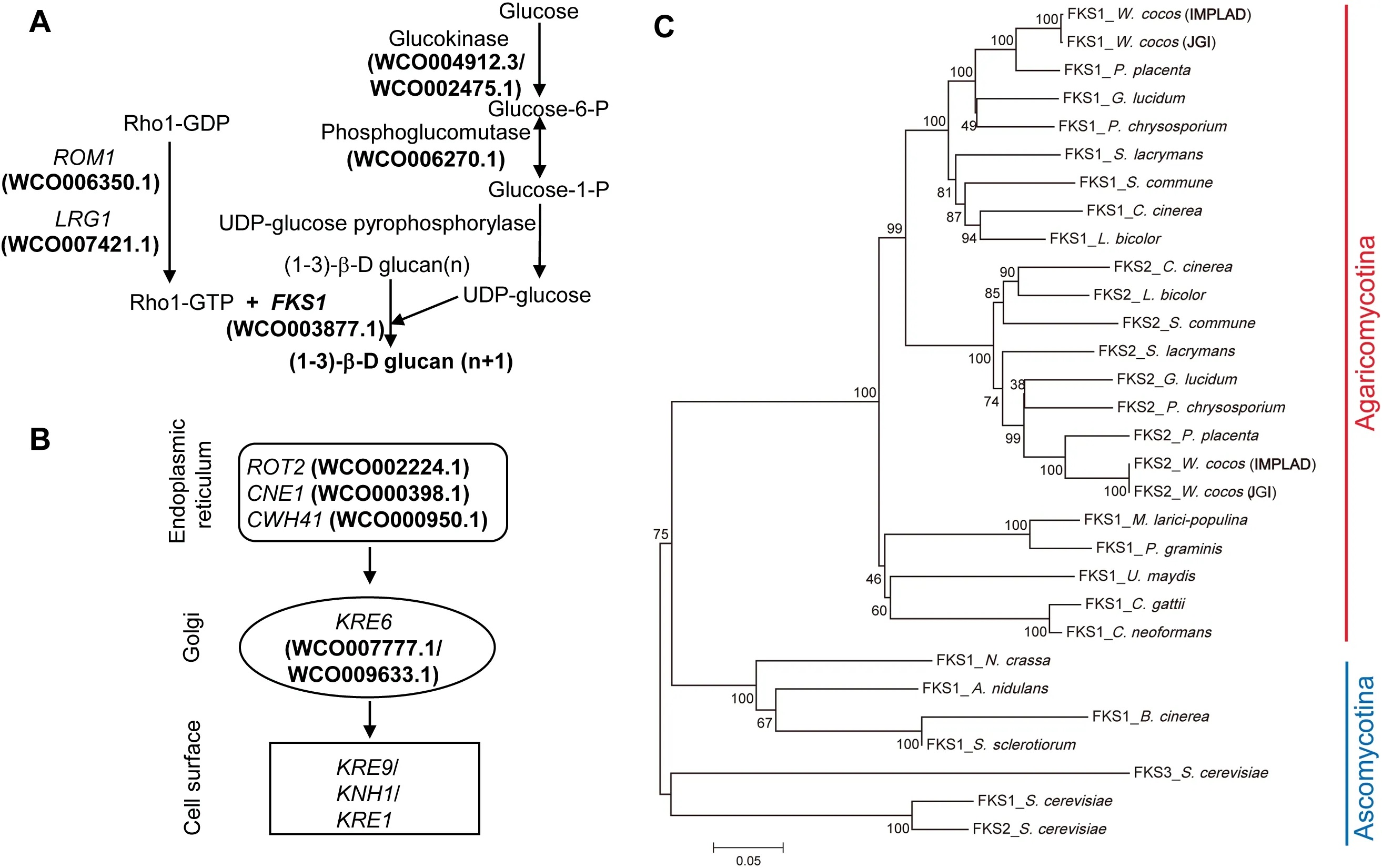
Figure 4 The putative biosynthetic genes and regulatory components of (1-3)-β-D and (1-6)-β-D glucan (polysaccharide) biosynthesisA.The predicted biosynthetic genes involved in(1-3)-β-D glucan synthesis in W.cocos.B.The putative biosynthetic steps involved in(1-6)-β-D glucan synthesis in W. cocos. C. The phylogeny of glucan synthase (FKS) proteins in W. cocos and other fungi, including of W.cocos(IMPLAD),W.cocos(JGI),P.placenta,P.chrysosporium,G.lucidum,S.lacrymans,S.commune,L.bicolor,C.cinerea,P.graminis,M.larici-populina,U.maydis,C.neoformans,C.gattii,N.crassa,A.nidulans,Sclerotinia sclerotiorum,Botrytis cinerea,and Saccharomyces cerevisiae. The two FKSs (FKS1 and FKS2) identified from W. cocos sequenced by IMPLAD and JGI, respectively, showed the highest homology among all the FKSs.ROM1,RHO1 multicopy suppressor;RHO1,Ras homologous 1;LRG1,LIM-RhoGAP homologous gene 1; FKS1, FK506-supersensitive 1; ROT2, reversal of TOR2; CNE1, calnexin; CWH41, calcofluor white hypersensitive 41; KRE6, killer toxin resistant 6; KRE9, killer toxin resistant 9; KNH1, KRE9 homolog; KRE1, killer toxin resistant 1.
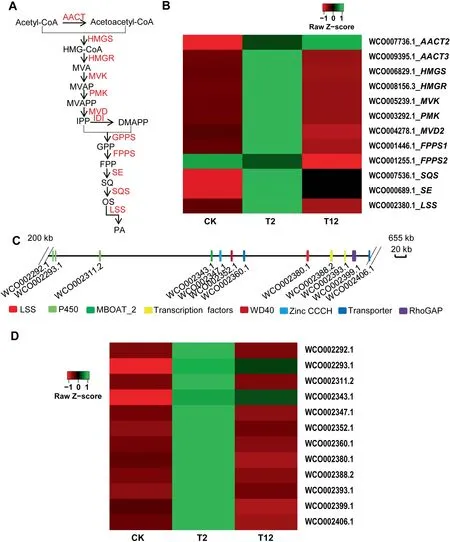
Figure 5 Biosynthesis of triterpenoids in W. cocosA.The putative biosynthetic pathway of PA,a lanostane-type triterpenoid in W.cocos with diverse bioactivities that is biosynthesized via the MVA pathway. All the genes encoding the predicted enzymes invovled in the MVA pathway have been identified in W. cocos(IMPLAD)genome.B.The expression patterns of the genes encoding enzymes involved in the MVA pathway induced by MeJA(200 μM)treatment for 2 h (T2) and 12 h (T12) compared to the control group (CK). C. The potential gene cluster including 12 genes for triterpenoid biosynthesis identified in W. cocos (IMPLAD) genome. These genes encoded P450s, transcription factors, transporters, and LSS.D.Coexpression analysis of the genes belonging to the LSS gene cluster induced by MeJA(200 μM)treatment for 2 h(T2)and 12 h(T12) compared to the control group (CK). AACT, acetyl-CoA acetyltransferase; HMGS, hydroxymethylglutaryl-CoA synthase A;HMGR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; MVK, mevalonate kinase; PMK, 5-phosphomevalomate kinase; MVD,diphosphomevalonate decarboxylase; IDI, isopentenyl diphosphate isomerase; GPPS, geranyl diphosphate synthase; FPPS, farnesyl diphosphate synthase; SE, squalene monooxygenase; SQS, squalene synthase; LSS, lanosterol synthase; PA, pachymic acid.
Lanosterol is the first cyclic intermediate for PA biosynthesis and is catalyzed by the rate-limiting enzyme LSS.A potential gene cluster involved in triterpenoid biosynthesis was located in scaffold 16 (Figure 5C; Table S29). The expression of 22 genes belonging to this cluster was measured in the mycelium incubated in the presence of MeJA (Table S29). Two CYP450 genes (WCO002292.1 and WCO002311.2), two TF genes (WCO002388.2 and WCO002393.1), one CCCH Zinc finger gene (WCO002347.1), one WD40 domain containing gene (WCO002352.1), and two transporter genes(WCO002360.1 and WCO002406.1) were significantly coexpressed with LSS (WCO002380.1) (Figure 5D), providing support for the predicted function of this gene cluster in triterpenoid biosynthesis in W. cocos.
Genes related to other secondary metabolites
Genome mining revealed multifunctional modular polyketide synthases (PKSs) and nonribosomal peptide synthetases(NRPSs) that are responsible for the biosynthesis of most biologically-active metabolites in microorganisms [34].Although previous studies have not identified any products of PKSs or NRPSs in W.cocos[3,35,36],a total of eight genes encoding NRPS-like proteins, seven genes encoding PKS proteins, and eight genes encoding PKS/NRPS-like proteins were identified in the present study(Table S30A).Similarly,13 predicted gene clusters containing these core genes were predicted by antiSMASH annotation[37](Table S30B and C),including five NRPS-like gene clusters and eight PKS gene clusters.These gene clusters consisted of 3–18 genes, implying that some clusters are incomplete.In addition,15 terpene gene clusters were also discovered(Table S30B and C).These gene clusters might participate in the production of terpenes.
Genes related to cell wall degradation
To identify enzymes involved in cell wall degradation, W.cocos genes were searched against the carbohydrate-active enzymes(CAZymes)database[38].A total of 321 genes encoding CAZymes were predicted in the W. cocos (IMPLAD) genome,including 139 glycoside hydrolase(GH)genes,63 glycosyl transferase (GT) genes, 58 carbohydrate esterase (CE) genes,20 carbohydrate-binding module (CBM) genes, 38 auxiliary activity (AA) genes, and 3 polysaccharide lyase (PL) genes(Table S31A). Most CEs, GHs, and AAs are involved in the hydrolysis of carbohydrate and noncarbohydrate substrates and the oxidative degradation of lignin-based components of the plant cell wall.
The presence of multiple gene copies for GH5, GH16,GH18, and GH28, which were involved in the degradation of cellulose, β-glucan, chitin, and pectin, respectively, and the absence of GH11 (encoding a xylanase) and CE12 (encoding an acetylesterase) in W. cocos (IMPLAD) are consistent with a previous study[39].The family 1 carbohydrate binding modules (CBM1), which is generally absent in brown-rot fungus,was also absent in W. cocos (IMPLAD). Compared with P.placenta and Fomitopsis pinicola, the gene numbers of GHs in both W. cocos (IMPLAD) and W. cocos (JGI) were similar to those in P. placenta but less than that of F. pinicola(Table S31B and C). CEs hydrolyze a wide range of carbohydrate and noncarbohydrate substrates. The W. cocos(IMPLAD) genome covered a higher number of CE family(58 genes)compared to other brown-rot species of fungi in this study.In addition,83 CAZyme genes were upregulated and 48 genes were downregulated in sclerotium compared to those in mycelium (Table S31A). The most abundantly upregulated genes in sclerotium were in the CE10 family(7 genes),followed by the GH16 (5 genes), GH13 (5 genes), and GH5 (4 genes)families.
Genes encoding membrane transporters
A total of 3710 transporters,belonging to 85 families,was predicted in W.cocos(IMPLAD),accounting for 34%of the total predicted genes (Table S32). The ATP-binding cassette (ABC)transporters were the most abundant category, together with 94 major facilitator superfamily (MFS) transporters, which may play key roles in the transportation of secondary metabolites in W.cocos.A total of 162 upregulated and 165 downregulated genes were detected in sclerotium compared to mycelium,indicating more active transportation in cells during sclerotial development and formation. It is worth noting that most of the significantly upregulated genes in sclerotium were members of the ABC gene superfamily, whereas most of the downregulated genes encode glycoside-pentoside-hexuronide(GPH)proteins,a cation symporter family,suggesting the different functions of these transporters in W. cocos.
Regulation of secondary metabolism and fungal development
Velvet protein family
The velvet protein complex plays essential roles in the regulation of fungal development and secondary metabolite production in Fungi [40,41]. Ten genes encoding velvet domaincontaining proteins were identified in W. cocos (Figure S6;Table S33). In particular, VosA (WCO008289.1) and VelB(WCO005599.1) showed significantly upregulated expression patterns in sclerotium. Several velvet genes were distributed in clusters with a head-to-tail structure, such as WCO009956.3–WCO009957.1,WCO000954.1–WCO000955.1,and WCO010849.1–WCO010850.1. All these velvet proteins have homologs in W. cocos (JGI) (Table S33).
Transcription factors
In total, 307 genes encoding TFs were predicted in W. cocos(IMPLAD) genome, with the most abundant family being the zinc knuckle TF family (130 genes), followed by the fungal-specific TF-domain family (43 genes) and the fungal Zn(2)-Cys(6)binuclear cluster domain family (29 genes)(Table S34). 69 TFs were differentially expressed between the transcriptomes of sclerotium and mycelium, including prominent TF genes in the fungal-specific TF-domain family and the fungal Zn(2)-Cys(6)binuclear cluster domain family, suggesting the suspected roles of these TFs in the regulation of fungal development in W. cocos.
Discussion
Herein,we characterized the genome and transcriptome of the Chinese strain CGMCC5.78 of W. cocos to investigate the molecular mechanisms related to the formation of the edible and medicinal sclerotium. We identified several differentially expressed genes (DEGs) that may be involved in sclerotial development and characterized metabolic pathways that are involved in the biosynthesis of several bioactive secondary metabolites, such as triterpenoids and glucans.
The present comparative genome analysis also uncovered substantial genetic variations between the Chinese(CGMCC5.78) and American (MD-104 SS10) strains of W.cocos(Figure 2,Figure S2),which are consistent with the polymorphism observed between another Chinese and a Japanese W. cocos strains [42]. Transcript profiling of the vegetative mycelium and sclerotium (Table S15) showed that a striking metabolic reorganization is induced by the mycelium-tosclerotia transition. The AS events (Figure 3B; Tables S19 and S20) and novel TUs (Table S21) provide useful information for further research on gene characterization in W.cocos.
The molecular basis of sclerotial formation has been elaborated in S. sclerotiorum [9,10]. W. cocos possesses a similar genomic capacity for the formation of its edible and medicinal sclerotium. Sclerotial development comprises multiple processes and is affected by environmental changes(e.g.,oxidative stress and low pH),primary metabolism(e.g.,fatty acid desaturation and degradation pathways), secondary messengers,and signaling components [9–12,43,44]. Large sclerotia of W.cocos can be induced by an early inoculation of small sclerotia on the medium [45], indicative of a specific signal transmitted from the small sclerotia for the induction of larger sclerotia.Signaling factors (e.g., pH-responsive TFs, Pac1, heterotrimeric G proteins, MAP kinases, Ras, protein phosphatases,and adenylate cyclases) might be responsible for the revulsive cultivation of sclerotia (Table S22). The principal difference in the genomic repertoire involved the expansion of the heterotrimeric G protein subunits (e.g., Gα subunit) and the monomeric GTPase modules (e.g., RhoGEF proteins), which represent the candidate sclerotium-associated factors in W.cocos.
Sclerotial formation begins with the degradation of plant cell walls and relies on the colonization of woody tissues[46]. A sustainable carbon source is one of the nutritional requirements for sclerotial development [39]. W.cocos decomposes celluloses and hemicelluloses of Pinus species early in the decay process,similar to other brown wood-decaying fungi,to provide the carbon required for substantial sclerotial development and formation [46]. CAZymes (Table S31) contribute to the degradation of pine wood tissues and release carbohydrates and other unknown nutrients that are absorbed and utilized by W. cocos to form sclerotia [39]. W. cocos genome is enriched in CE genes (58 genes) (Table S31). This CAZyme repertoire differs from other brown-rot fungi and may be related to the specific habitat of W. cocos.
Previous studies showed that the mating type of W. cocos appears to be bipolar [47,48], and the primary and secondary hyphae are without clamp connections [46,48]. In the mating process, HD genes and pheromone receptor genes are physically linked and localized in one mating-type locus (MAT-A locus) in bipolar species. In tetrapolar species, these two types of genes are located in two unlinked loci and designated as the MAT-A (or HD) locus and MAT-B (or pheromone receptor gene)locus[49,50].The MAT-A locus genes are organized with a highly conserved gene order that includes one or more pairs of HD1 and HD2 [51], with an MIP and a β-FG flanking the HD1-HD2 gene pair(s) [47]. The HD1-HD2 gene pair of W.cocos was located in the same scaffold with an MIP, while the β-FG was found in another scaffold (Figure S4A;Table S24). This may result from the assembly fragmentation.The HD and pheromone receptor genes are present in all Agaricomycete genomes, regardless of whether they are bipolar or tetrapolar[49].Consequently,two STE3-like pheromone receptor genes (WCO006868.1 and WCO007080.1) belonging to the MAT-B locus were also identified in the W. cocos genome (Table S24). These mating locus genes provide molecular evidence for the determination of the mating process of W.cocos.
The biosynthesis of secondary metabolites is often coupled with morphological development in fungi[52].Several polysaccharides and triterpenoids are bioactive compounds that are responsible for the medicinal function of W. cocos. Bioactive polysaccharides comprise dietary fibers accumulating in sclerotium, including a structurally diverse class of biological macromolecules in the cell wall with wide-ranging physicochemical properties[3].Linear(1-3)-β-D glucan is the primary constituent of the polysaccharides isolated from the W. cocos sclerotium [53]. Because molecular components involved in the biosynthesis and regulation of polysaccharides in cell wall have been characterized in yeast [28,29], we were able to identify the homologous genes in W. cocos (Figure 4; Table S26),providing the most likely candidate genes invovled in polysaccharide biosynthesis and regulation in W. cocos.
Although several MVA pathway-related genes have been identified in W. cocos [21], the present W. cocos genome provides the entire collection of genes and a potential gene cluster participating in PA and triterpenoid biosynthesis (Figure 5;Table S28). The gene cluster is similar to a typical terpene biosynthetic gene cluster; it contains the LSS signature gene that is clustered with genes coding for TFs and transporters that likely correspond to tailoring enzymes and components(Figure 5C; Table S29) [54,55]. Moreover, most of the genes in this cluster were coexpressed with LSS after MeJA induction, which conforms to the characteristics of a gene cluster(Figure 5D). Intriguingly, this gene cluster has not been identified in the G.lucidum genome[56],inferring the specific function of the gene cluster for PA biosynthesis in W. cocos.
The expression of biosynthetic genes related to secondary metabolites is governed by a network of key regulators that respond to diverse environmental cues. The velvet proteins, a class of key regulators, can coordinate the crosstalk of secondary metabolism and differentiation processes,such as asexual or sexual sporulation and sclerotium or fruiting body formation [41]. In W. cocos, the velvet gene family (ten members) is expanded compared to B. cinerea, S. sclerotiorum, P.chrysosporium, and G. lucidum (Table S33). This gene expansion might be due to tandem duplication of velvet genes in W.cocos.In addition,the signaling components of G proteins and other regulators (including TFs) (Table S34) also regulate the crosstalk of secondary metabolism and fungal development[52,57].This molecular information will be helpful in the illustration of the regulatory mechanisms controlled by signaling components in W. cocos.
Conclusion
The present W.cocos genome and transcriptome resources will provide novel opportunities for investigating the mechanisms driving the formation of edible and medicinal sclerotia in fungi. This study also provides new insights into the diversification of the molecular mechanisms involved in fungal development and secondary metabolism in W. cocos. We identified several DEGs related to signaling components, secondary metabolism, nutrient transport, TFs, mating process,and CAZymes that are likely involved in the sclerotial formation (Figure S7). Finally, we have identified several genes involved in the biosynthesis of key secondary metabolites that are relevant to the medicinal properties of the fungus.
Materials and methods
Fungal strain and culture conditions
W. cocos dikaryotic strain (CGMCC5.78) was obtained from the China General Microbiological Culture Collection Center(Beijing,China,http://www.cgmcc.net/)and stored in our laboratory. This is one of the most widely used strains in China.Vegetative mycelia were grown on potato dextrose medium in the dark at 28°C.Liquid cultures for the mycelia were shaken at 50 rpm for 7 days. The mature sclerotia of the strain CGMCC5.78 used for transcriptome analysis were cultivated on Quercus variabilis Blume logs at HuiTao Pharmaceutical Company (Luotian, China).
Construction of the fosmid library
High-molecular-weight(HMW)DNA was isolated from 7-day cultured mycelia using the CTAB DNA isolation method.Genomic DNA was randomly sheared to approximately 40 kb in size. Then, the sheared molecules were size-selected using Pulse Field Gel Electrophoresis (Bio-Rad, Hercules,CA).The sheared DNA was ligated into the pCC2FOS vector(Epicentre, Madison, WI) after end-repair. The ligations were packaged using MaxPlax Lambda Packaging Extracts (Epicentre). The lambda phages carrying foreign DNA were used to infect EPI300-T1R E. coli cells (Epicentre). Positive fosmid clones were selected using lysogeny broth (LB)-chloramphenicol (12.5 μg/ml) plates. A total of 11,232 clones were selected and transferred to 96-well plates containing frozen LB media and stored at -80 °C.
De novo genome sequencing and assembly
A strategy combining fosmid construction, fosmid shotgun sequencing, and assembly was used in this study. In total, we obtained 11,232 fosmid clones from the fosmid library.Different indices and adapters were added to the fosmid clones to construct the sequencing library. The clones were pooled and sequenced by using HiSeq 2000 (Illumina, San Diego, CA).Finally, we obtained 178.44 Gb data. In addition, we constructed genomic libraries with 511 bp, 2 kb, 5 kb, 10 kb,and 20 kb in length and used the paired-end information to construct contigs and longer scaffolds.A set of exclusion steps was included following preprocessing.These steps included the removal of reads with low-quality bases>40 bp or reads with N base content of more than 10%or poly A.The overlapping reads (read1 and read2 overlap at least 10 bp and mismatch less than 10%) and duplicate reads (the sequence of read1 was equal to read2, which means duplication) were also excluded. After removal of the vector sequence from fosmid clones, the reads generated from the HiSeq 2000 were used in assembly. The short reads were first assembled into fosmid clone sequences using SOAP de novo [24]. After removing the remaining vector sequences and other low-quality bases, the fosmid clone sequences were assembled into contig sequences using Celera software. The redundant sequences were first removed via BLAST by alignment to the National Center for Biotechnology Information (NCBI) microbial nucleotide database for any alignment query contigs.Then,the redundant sequences based on the K-mer frequencies obtained from the short library reads were removed.Finally,the contig sequences were connected into scaffold sequences by using SSPACE(version 1.1)software.The assembled contig sequences were manually corrected to build the finished scaffolds.
Gene prediction and annotation
De novo gene prediction was executed using Augustus,GeneMark-ES, and SNAP based on the Hidden Markov Model (HMM). Additionally, the homolog-based method was used to weight and calibrate gene structure.The combination of de novo gene prediction and gene structure prediction was then used to obtain the GLEAN gene groups. The GLEAN data were corrected by RNA-seq data to accomplish gene prediction.Genes were functionally predicted by BLAST against the public databases, including NCBI nonredundant protein database (Nr), NCBI nonredundant nucleotide database (Nt), InterProScan, GO database, KEGG database,Clusters of Orthologous Groups (COG) database, and TrEMBL/Swissprot database. The PFAM families of TFs in fungi were selected to search the W. cocos genome [58]. The Transporter Classification Database (TCDB; http://www.tcdb.org) [59] were used to designate transporters in the W. cocos genome by BLAST (E-value = 1E–10).
Identification of TEs and noncoding RNA
TEs were searched in the W. cocos genome using RepeatMasker and ProteinMask software based on Repbase(16.03)data.The de novo genome repeat sequence database was then constructed by PILER-DF, RepeatScout, and LTR-Finder, and redundant sequences were removed. TEs were identified with the RepeatMasker software in the clean de novo genome repeat sequence database. tRNAs were identified by tRNAscan-SE software.rRNAs were identified by BLAST against the rRNA database of S. commune.
Ortholog analysis
Orthologous groups from the related fungal genomes of Coprinopsis cinerea, Cryptococcus gattii, Cryptococcus neoformans, Aspergillus nidulans, L. bicolor, Melampsora laricis-populina,Neurospora crassa,P.chrysosporium,Puccinia graminis, S. cerevisiae, S. commune, Serpula lacrymans, and Ustilago maydis were identified using OrthoMCL [25] version 2.0 (http://www.orthomcl.org). The cutoff of BLASTP was set as E-value < 1E–5.
Transcriptome sequencing and analysis
Transcriptome analyses were performed on the materials obtained from the 7-day-old mycelia and mature sclerotia of W. cocos. Frozen samples were ground to powder in liquid nitrogen for RNA isolation. Total RNA from each sample was extracted using an RNeasy Plant Mini Kit (Qiagen,Dusseldorf, Germany) according to the manufacturer’s instructions. These total RNA samples were also reversetranscribed into cDNA to validate the RNA-seq data by RT-qPCR method. Genomic DNA was eliminated from the total RNA using DNase I (NEB, Ipswich, MA). RNA integrity and quality were tested using the RNA 6000 Nano II kit on a Bioanalyzer 2100 (Agilent Technologies, Lexington,MA). The RNA samples were sequenced using an Illumina HiSeq 2000 (Illumina). RNA-seq data from the mycelial and mature sclerotial tissues were assembled into transcripts using Trinity. Transcript abundance was estimated by the RPKM method [60]. Differential expression analysis was executed according to the method reported by Audic and Claverie[61]. The AS events were analyzed by TopHat software [62].
RT-qPCR analysis of gene expression
Total RNA was extracted from 10-day-old W. cocos mycelia under the treatment of 200 μM MeJA for 0 h (CK), 2 h(T2), and 12 h (T12), respectively. The RNA was then treated with recombinant DNase I (Life Technologies, Burlington,Canada) at a concentration of 1.5 U/μg total RNA. Singlestrand cDNA was synthesized using the PrimeScriptTM1stStrand cDNA Synthesis kit (TaKaRa, Shlga, Japan), 1 μg RNase-free DNase I-treated (TaKaRa) total RNA, and random primers. RT-qPCR was performed at least three times for each sequence using SYBR?Premix Ex TaqTM (Perfect Real Time) (TaKaRa) and an ABI PRISM 7500 real-time PCR System (Life Technologies). Each reaction contained 7.5 μl 2× SYBR Green Master Mix Reagent (Life Technologies), 1.0 μl (10 ng) cDNA, and 200 nM gene-specific primers in a total reaction volume of 15 μl.The PCR amplification program consisted of denaturation at 95°C for 30 s followed by 40 cycles of 95 °C for 5 s and 60 °C for 34 s. The relative gene expression data were normalized against an internal reference gene, cyclophilin (CYP) [23]. The relative expression levels were calculated by the 2-ΔΔCtmethod. The primers were designed using Primer3 (http://frodo.wi.mit.edu/primer3/).The primers used for RT-qPCR in this study are listed in Table S35.
Data availability
The raw data for the genome and transcriptome of the Chinese strain CGMCC5.78 W. cocos have been deposited in the Genome Sequence Archive [63] at the National Genomics Data Center,Beijing Institute of Genomics,Chinese Academy of Sciences/China National Center for Bioinformation(GSA:CRA003688), and are publicly accessible at http://bigd.big.ac.cn/gsa.
CRediT author statement
Hongmei Luo:Writing-original draft,Writing-review&editing, Formal analysis. Jun Qian: Software, Visualization. Zhichao Xu: Formal analysis. Wanjing Liu: Validation, Data curation. Lei Xu: Resources, Investigation. Ying Li: Formal analysis. Jiang Xu: Investigation, Data curation. Jianhong Zhang: Validation, Data curation. Xiaolan Xu: Validation,Data curation. Chang Liu: Formal analysis. Liu He: Validation, Data curation. Jianqin Li: Formal analysis. Chao Sun:Formal analysis. Francis Martin: Writing - review & editing.Jingyuan Song: Supervision, Methodology. Shilin Chen: Conceptualization, Funding acquisition. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (Grant No. 2016-I2M-3-016) and the National TCM Standardization Project (Grant No.ZY13ZH-C-JL-24). Francis Martin is funded by the French National Research Agency through the Laboratory of Excellence Advanced Research on the Biology of Tree and Forest Ecosystems (Grant No. ANR-11-LABX 0002 01) and Beijing Advanced Innovation Center for Tree Breeding by Molecular Design, Beijing Forestry University. We thank the Beijing Genomics Institute (BGI) for the project management that generated the initial sequence and assembly. We also thank Yongchao Niu and Jinmin Lian(Biozeron Co.Ltd,Shenzhen,China) for the support in data analysis.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.01.007.
ORCID
0000-0002-3403-7659 (Hongmei Luo)
0000-0001-5923-0538 (Jun Qian)
0000-0003-1753-5602 (Zhichao Xu)
0000-0002-6485-8319 (Wanjing Liu)
0000-0002-4841-630X (Lei Xu)
0000-0001-9560-2212 (Ying Li)
0000-0002-1139-4063 (Jiang Xu)
0000-0002-8394-1951 (Jianhong Zhang)
0000-0001-8476-3643 (Xiaolan Xu)
0000-0003-3879-7302 (Chang Liu)
0000-0003-3951-4132 (Liu He)
0000-0003-3792-1172 (Jianqin Li)
0000-0001-7096-5033 (Chao Sun)
0000-0002-4737-3715 (Francis Martin)
0000-0003-2733-0416 (Jingyuan Song)
0000-0002-0449-236X (Shilin Chen)
 Genomics,Proteomics & Bioinformatics2020年4期
Genomics,Proteomics & Bioinformatics2020年4期
- Genomics,Proteomics & Bioinformatics的其它文章
- Global Analysis of Gene Expression Profiles Provides Novel Insights into the Development and Evolution of the Large Crustacean Eriocheir sinensis
- mrMLM v4.0.2: An R Platform for Multi-locus Genome-wide Association Studies
- Denoising Autoencoder, A Deep Learning Algorithm, Aids the Identification of A Novel Molecular Signature of Lung Adenocarcinoma
- G9a/GLP-sensitivity of H3K9me2 Demarcates Two Types of Genomic Compartments
- Tissue-specific Gene Expression Changes Are Associated with Aging in Mice
- Transcriptomic and Proteomic Analysis of Mannitol-metabolism-associated Genes in Saccharina japonica