
SinoDuplex:An Improved Duplex Sequencing Approach to Detect Low-frequency Variants in Plasma cfDNA Samples
2020-07-29 05:34:46YongzheRenYangZhangDandanWangFengyingLiuYingFuShaohuaXiangLiSuJianchengLiHengDaiBingdingHuang
Yongzhe Ren ,Yang Zhang ,Dandan Wang ,Fengying Liu ,Ying Fu ,Shaohua Xiang ,Li Su ,Jiancheng Li ,Heng Dai ,Bingding Huang ,,5,*
1College of Big Data and Internet,Shenzhen Technology University,Shenzhen 518118,China
2Department of Research and Development,Sinotech Genomics Inc.,Kanxing Road 3399,Shanghai 201314,China
3Department of Integrated Traditional and Western Medicine In Oncology,The First Affiliated Hospital of Anhui Medical University,Hefei 230022,China
4Department of Radiation Oncology,Fujian Medical University Cancer Hospital and Fujian Cancer Hospital,Fuzhou 350014,China
5 Institute of Synthetic Biology,Shenzhen Institute of Advanced Technology,Chinese Academy of Science,Shenzhen 518005,China
KEYWORDS Next generation sequencing;Liquid biopsy;Circulating tumor DNA;Duplex sequencing;Low frequency variant
Abstract Accurate detection of low frequency mutations from plasma cell-free DNA in blood using targeted next generation sequencing technology has shown promising benefits in clinical settings.Duplex sequencing technology is the most commonly used approach in liquid biopsies.Unique molecular identifiers are attached to each double-stranded DNA template,followed by production of low-error consensus sequences to detect low frequency variants.However,high sequencing costs have hindered application of this approach in clinical practice.Here,we have developed an improved duplex sequencing approach called SinoDuplex,which utilizes a pool of adapters containing pre-defined barcode sequences to generate far fewer barcode combinations than with random sequences,and implemented a novel computational analysis algorithm to generate duplex consensus sequences more precisely.SinoDuplex increased the output of duplex sequencing technology,making it more cost-effective.We evaluated our approach using reference standard samples and cell-free DNA samples from lung cancer patients.Our results showed that SinoDuplex has high sensitivity and specificity in detecting very low allele frequency mutations.The source code for SinoDuplex is freely available at https://github.com/SinOncology/sinoduplex.
Introduction
Liquid biopsies are valuable tools for non-invasive diagnostics and monitoring of diseases such as cancer.These can include sampling of peripheral blood,effusion fluids,and other components of body fluids for extraction of circulating tumor cells(CTCs)[1],tumor-derived cell-free DNA(cfDNA),and other materials(e.g.,exosome,extracellular vesicles)[2].Although PCR-based testing is recommended for routine diagnostics of cancer hotspot mutations [3,4], targeted next-generation sequencing(NGS)using cfDNA,short DNA fragments in the plasma released by apoptotic and necrotic cells, has emerged as the most common approach to assess tumorspecific alterations[5,6].For instance,it has been used to determine the genetic landscape of tumor lesions,monitor treatment responses,track acquired resistance,select existing antiresistance targeting therapies,and assess the presence of residual disease [1,4]. Compared to tissue biopsies, targeted sequencing in liquid biopsies currently appears to be a highly promising and revolutionary tool for diagnosing and monitoring cancer.The method has three major advantages:(i)liquid biopsies enable repeated sampling over a relatively long period of time to monitor the patient’s clinical condition,whereas tissue biopsies cannot be obtained repeatedly or be taken from a specific cancerous lesion location;(ii)liquid biopsies overcome single-biopsy bias,enabling representation of the full extent of tumor heterogeneity;and(iii)PCR-based assays can only detect one or a few known genomic hotspot mutations,which may only apply to a minority of patients.In Asian patients,for example,47%of lung cancer patients had EGFR mutations,compared with only 15%in European patients[7].
Targeted NGS technology can detect genetic alterations in a wider pool of genomic regions,and has been employed to screen rare mutations in early-stage cancers[8,9],guide targeted therapy,monitor treatment,and enable prognoses[1].However, sub-clonal genetic mutations in tumors may be found in less than 1%of DNA molecules in a plasma sample[10,11].In addition,cfDNA extracted from blood samples usually contain damaged DNA from normal metabolic processes or as a result of DNA extraction[12-14].Consequently,these artefactual mutations might be retained during the PCR amplification process.Additionally,mutations may be misincorporated by DNA polymerase during PCR amplification[15-17]. Therefore, additional refinement for ultrasensitive profiling of circulating tumor DNA(ctDNA)is essential to improve ctDNA quantity and quality,and reduce errors introduced during PCR amplification,library construction and sequencing.To improve sensitivity and minimize errors,different approaches have been developed using unique molecular identifiers(UMIs or molecular barcodes).This allows each DNA molecule to be labeled and accurately tracked.The first barcode strategy was developed to track single DNA strands[18-22].Recently,duplex sequencing(DS),an improved barcoding strategy for tracking double-stranded DNA,was also reported[10,21].DS uses randomly generated barcodes to uniquely tag each DNA fragment in a plasma cfDNA sample.Tagged fragments are then amplified by PCR before being used in the preparation of a sequencing library,creating fragment families characterized by unique combinations of barcodes at both the 5′and 3′ends.A family contains multiple reads,each originating from a single input DNA fragment.A true variant will appear in all reads within a family.In contrast,sequencing and amplification errors will manifest themselves as‘‘polymorphisms”within a family,thus allowing them to be identified and removed by generating consensus sequences.The consensus of all reads originating from the same strand reduces errors originating from PCR amplification and sequencing.Only mutations present in sequences obtained from both complementary DNA strands are counted as true positive mutations. The sequences are referred as duplex consensus sequences(DCS),whereas mutations present in only one of the complementary DNA strands(single-strand consensus sequences,SSCS)are still counted as errors.
Although DS is expected to drive significant advances and improve sensitivity in detecting rare mutations in ctDNA,the experimental and computational aspects of this technique are still evolving[22-25].In this study,we have developed a novel,efficient DS approach named SinoDuplex,combining a special barcoding strategy consisting of pre-defined barcode sequences with a novel computational algorithm to eliminate background noise and produce more accurate duplex consensus sequencing data.We evaluated the performance of Sino-Duplex with a pool of diluted samples using two reference standard samples,HD701 and HD753,on a targeted panel of 334 genes.SinoDuplex increases the output of DS while reducing cost.At an allele frequency cut-off of 0.1%,SinoDuplex achieved a high sensitivity of 98.62%and specificity of 97.09%.In addition,we applied this method to samples from patients with clinical lung cancer and validated low-frequency hotspot actionable mutations with droplet digital PCR(ddPCR). Our results show that SinoDuplex significantly improves the sensitivity and specificity for the detection of low-frequency variants in plasma ctDNA samples in a costeffective manner.
Materials and methods
SinoDuplex adapter synthesis
Our novel duplex adapters (termed SinoDuplex adapters)employ a pool of at least 16 unique molecular identifiers(UMIs)at the end of the double-strand portion of the Yshaped adapters.The pool of UMIs comprises seven or eight bases of pre-defined and color-balanced sequences mixed in certain ratios to avoid sequencing bias on Illumina sequencing instruments.Every pre-defined UMI differs from all other UMIs by at least three edit distances.SinoDuplex adapters are formed by combining and annealing two single strands of oligonucleotides possessing pre-defined UMI in a pairwise manner.One oligonucleotide is designated as the P5 strand:ACACTCTTTCCCTACACGACGCTCTTCCGATCTXXX XXXX(X)T(where XXXXXXX(X)indicates the position of a fixed 7-or 8-base UMI sequence),and the other is designated as the P7 strand:/5phos/X‘X’X‘X’X‘X’X‘(X’)AGAT CGGAAGAGCACACGTCTGAACTCCAGTCAC (where X‘X’X‘X’X‘X’X‘(X’) indicates the reverse complement of XXXXXXX(X)).(Table S1).Each pair of adapter strands was synthesized(HPLC and NGS grade,Life Technologies,Carlsbad,CA)and combined by equimolar amounts to a final concentration of 100 μM in 1×annealing buffer containing 10 mM Tris,1 mM EDTA and 0.1 M NaCl.Each reaction was heated to 95 ℃for 5 min in an ABI 9700 thermocycler(Applied Biosystems,Foster City,CA)before turning off the machine and leaving the reaction to gradually cool for 1 h.The annealed adapters were finally pooled together in equal volumes and further diluted to 10 μM in Low TE buffer to form a working solution.
DNA reference sample preparation
To estimate the performance of our DS strategy,two wellcharacterized genomic DNA reference standard samples,HD701 and HD753 (Horizon Discovery Inc., Cambridge,UK)were used.These samples are commercially available mixtures of DNA from cell lines for which precise allelic frequencies(AFs)of several hotspot actionable mutations have been validated by digital PCR,covering a wide range of mutations including single nucleotide variants(SNVs),indels,fusions,and copy number variation.Allele frequencies of the verified variants in these references were between 1%and 41.5%(details of each variant are provided in Table S2).A dilution series of these two reference samples (HD701M1, HD701M2,HD753M1,and HD753M2)with the HapMap normal cell line NA18536 were generated to simulate different ranges of allele frequencies and assess the performance and limit of detection of our assay.Diluted DNA mixtures were further sheared using a focused-ultrasonicator(S220,Covaris,Woburn,MA)with a target size around 170 bp to mimic the size distribution of cfDNA.
Patient plasma samples and cfDNA extraction
A number of liquid biopsy specimens were collected to assist in validation experiments.For each sample,8-10 mL peripheral blood was collected in a cell-free DNA BCT tube(Catalog No. 218962, Streck, La Vista, NE) and centrifuged for 10 min at 1600g at room temperature within three days of drawing blood.The supernatant containing the plasma was further centrifuged at 16,000g for 10 min at room temperature to remove any residual cells.cfDNA was extracted from 4 to 5 mL of plasma and eluted into 50 μL of buffer AVE using the QIAamp Circulating Nucleic Acid Kit (Catalog No.5114,Qiagen,Hilden,Germany)according to manufacturer’s instructions. Quantification of extracted cfDNA was performed using the Qubit 3.0(Thermo Fisher Scientific,Waltham,MA).
Panel design
To apply SinoDuplex in a clinical setting,we designed three different targeted gene panels to detect low-allele-frequent mutations in plasma ctDNA samples.The smallest panel is a specially designed panel for lung cancer named LungCore which covers 10 core genes that have targeted drugs approved by the US FDA for lung cancer and actionable fusion events.The second panel,ActionAll,covers 73 genes with actionable mutations in targeted clinical therapy for all solid tumors.The third panel is a pan cancer panel covering the exons of 334 genes and is capable of estimating blood tumor mutational burden (TMB) to select cancer patients suitable for immunotherapy.The genes in this pan-caner panel are selected to cover variants associated with targeted cancer therapies(i)approved by the FDA or listed in the NCCN guidelines,(ii)reported as responsive to therapy in public databases and the literature. Hotspot actionable fusion introns were also included in these three panels to identify actionable fusion events.Capture probes were ordered from Integrated DNA Technologies,Coralville,IA.Gene lists and hotspot introns of these three panel are provided in Table S3.
Targeted library construction and sequencing
Pre-capture library preparation was performed using the KAPA Hyper Prep kit(Catalog No.K8504,Roche,Basel,Switzerland)with SinoDuplex adapters.In brief,20-33 ng cfDNA or sheared reference DNA mixture, representing 6000-10,000 haploid genomic equivalents,was used for end repair and A-tailing,followed by ligation of SinoDuplex adapters.Ligated products were bead-purified and further amplified for seven cycles using KAPA HiFi HotStart ReadyMix with unique dual indexes(UDIs),primers that mitigate sample mis-assignment due to index hopping. For each library,500 ng DNA with different UDIs were pooled together.Up to 2 μg of total library was used as input for in-solution capture enrichment with xGen Lockdown Reagents kit(Catalog No.1072281,Integrated DNA Technologies,Coralville,IA)and customized xGen lockdown panels.A hybridization mixture of pooled libraries and customized xGen lockdown probes in xGen Hybridization Buffer was denatured at 95°C for 5 min,then incubated at 65°C for 4-16 h with the addition of human Cot-1 DNA(Catalog No.15279011,Life Technologies,Carlsbad,CA)and xGen Universal Blockers-TS Mix(Catalog No.1075475,Integrated DNA Technologies,Coralville,IA).After incubation,library-probe duplexes were captured with Dynabeads M270 Streptavidin (Catalog No.65306,Invitrogen,Carlsbad,CA)and off-target library fragments were washed off.Bead-captured libraries were further amplified with universal P5/P7 primers in KAPA HiFi HotStart ReadyMix,followed by purification with beads.Postcapture libraries were quantified by Qubit 3.0.Fragment size was determined by a 2100 Bioanalyzer using a High Sensitivity DNA chip(Catalog No.5067-4626,Agilent Technologies,Santa Clara,CA).Paired-End 150 sequencing was performed using the HiSeq X Ten platform(Illumina,San Diego,CA)supporting dual indexing with raw sequencing depths over 20,000×.
Duplex consensus sequence generation
The complete computational workflow of the SinoDuplex approach is illustrated in Figure 1.Firstly,the QC and adapter trimming step is performed using fastp[26]to remove adapter contamination and filter out low-quality reads.Simultaneously,UMI barcode sequences are extracted from the read sequences and appended into the read name.Next,read pairs are mapped to the reference genome hg19 using the BWA mem algorithm[27]with‘‘-C”option to set the UMI barcode sequences as a special‘‘BC:Z”tag for each alignment in the bam file.Single-stranded consensus sequences(SSCS)are generated by loading all read pairs with the same mapping positions and orientations into memory and grouping them into different SSCS families with the same UMI barcode sequences at both ends and the same CIGAR string.Unlike the original method,which employs the majority-based algorithm[21],we apply a Bayesian algorithm to determine the final high-quality base at each position of the SSCS consensus sequence and calculate the corresponding consensus quality score for this base using the following Equation(1).
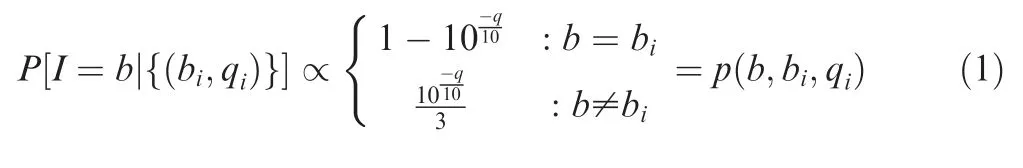
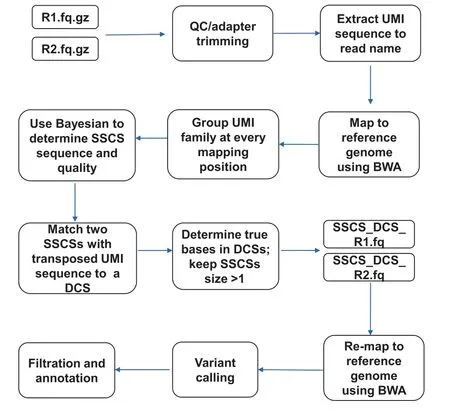
Figure 1 Schematic illustration of SinoDuplex workflow
Recall that each base b is associated with a base call and quality score pair(bi,qi)in the consensus group.The posterior probability of the consensus base given the entire consensus group is

The base with maximum posterior probability is taken as the consensus base and its corresponding quality score is calculated according to the chosen probability.
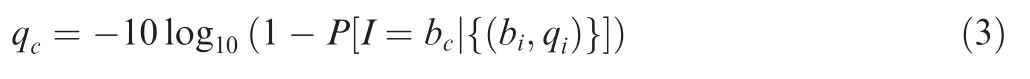
After construction of the SSCS sequence and quality score from all SSCS families for each mapping position,our algorithm merges two SSCS read pairs with transposed UMI barcode sequences and the same mapping position into one DCS,if possible.If both bases at the same position in two SSCSs(forward and reverse strands)match,the given base is used and an average Phred quality score is assigned to this base.However,in the case of a mismatch between two bases,the nucleotide‘‘N”is placed in final DCS sequence and a predefined low Phred score(10 is used here)is assigned.If the proportion of N bases in the final DCS sequence is greater than a pre-defined cut-off(50%in this study),this DCS sequence is filtered out.If the corresponding transposed read pair cannot be found for one SSCS,this SSCS read pair is also kept when its family size is greater than 1.The sequence and quality scores of all SSCS and DCS read pairs are written out into two intermediate FASTQ files to release memory. These DCS and SSCS read pairs are then re-mapped onto the human hg19 reference genome using the BWA mem algorithm after processing all raw read pairs using the algorithm above.A final bam file containing both SSCS and DCS read pairs is obtained for further variant calling.
Variant calling
For variant calling of ctDNA samples,we used an algorithm based on samtools mpileup[28]developed in-house to detect somatic SNVs and indels[29,30].Our calling algorithm can detect rare somatic mutations with a frequency as low as 0.1%.Briefly,many candidate SNVs/indels were identified in tumor samples with at least three reads and the required mapping quality and base quality score.After querying against a normal reference sample with filtering conditions, eligible mutations were categorized as either germline or somatic.In the calling process,a series of filters were applied on the raw SNV/indel calls,including noise estimation from known SNPs,strand bias filtering, and noise filtering from neighboring regions to ensure reliable variant detection.In our experience,most false positive variants originate from alignment errors and repeat regions.These variants can be removed using a blacklist containing common mistakes from a pool of normal samples. Final high-confidence variants (SNVs and small indels)were then annotated with UCSC RefSeq gene information,dbSNP[31],1K Genome[32],ExAC[33],GnomAD[33],COSMIC[34],and Clinvar[35]using SNPEff[36]and an inhouse database-annotation module based on HTSlib(http://www.htslib.org/)library.
Performance calculation
To evaluate the detection capability and limitation of our algorithm,we calculated the sensitivity or positive percentage agreement(PPA)and specificity or positive predictive value(PPV)using the confirmed somatic mutations in reference standards HD701 and HD753 with different dilution ratios using the HapMap normal cell line NA18536. Mutations called in two undiluted samples of HD701 and HD753 with a variant allele frequency >1%were treated as a true mutation data-set and were manually reviewed using the Integrative Genomics Viewer[37].The allele frequencies of some hotspot actionable mutations in these two reference samples were already confirmed by ddPCR assays by Horizon Discovery,Cambridge,UK.Figure S1 shows a high correlation between the AFs detected by SinoDuplex and AFs confirmed by ddPCR for those hotspot actionable mutations in undiluted samples of HD701 and HD753(true mutation data-set).Mutations detected in diluted samples that are also present in the true mutation data-set are treated as true positive(TP);those that are not,as false positive(FP).Mutation in the true mutation data-set that are not detected in the diluted sample are classified as false negative(FN).Thus,sensitivity and specificity are calculated as TP/(TP+FN)and TP/(TP+FP).Detailed information of all mutations(TP,FN,TP)for the diluted samples are provided in Table S4.To determine the appropriate sequencing depth,we performed in silico downsampling of diluted samples and calculated sensitivity and specificity at different depths using the same procedure.
Droplet digital PCR(ddPCR)validation
Hotspot actionable mutations such as KRAS(G12D)and EGFR(790M,L858R,and 19Dels)detected in patient cfDNA samples by SinoDuplex were further validated by ddPCR.In brief,two probes targeting mutant and wild-type alleles werelabeled with FAM and VIC dyes,respectively.A TaqMan PCR reaction using ddPCR Supermix for Probes(Catalog No.1863024,Bio-Rad,Hercules,CA)was subjected to droplet generation using the QX200 Droplet Digital PCR system(Bio-Rad,Hercules,CA),followed by PCR.Droplets were analyzed with the QX200 Droplet Reader(Bio-Rad,Hercules,CA)and QuantaSoft software(Bio-Rad,Hercules,CA)for fluorescent measurement and allele calling.
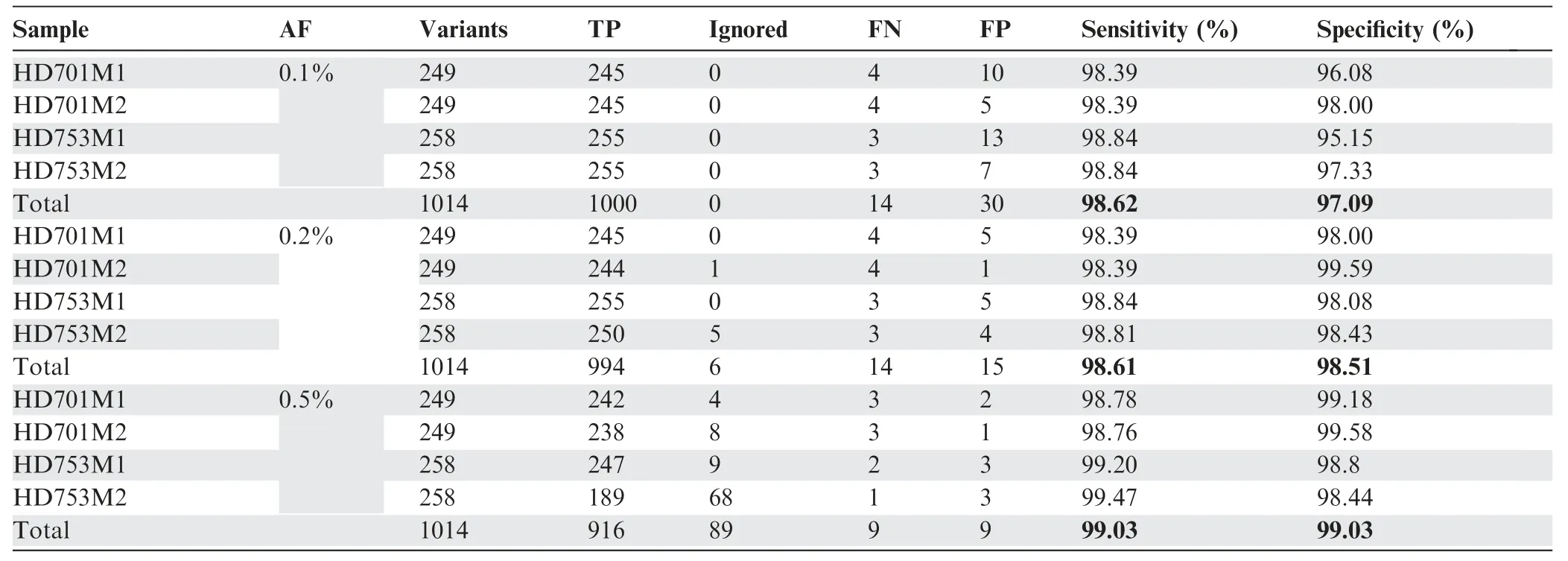
Table 1 The performance of SinoDuplex at different AF cut-offs:0.1%,0.2%,and 0.5%
Results
Improvement of low-frequency variant identification
To assess the performance of our approach(Figure 1)in detecting low-frequency variants,we calculated the sensitivity and specificity for a pool of diluted samples with known variants in the reference standard samples HD701 and HD753.To simulate a serial dilution of different allele frequency variants,HD701M1 and HD701M2 were diluted 3-fold and 6-fold with the HapMap normal sample NA18536. HD753M1 and HD753M2 were diluted 5-fold and 10-fold with NA18536.All diluted samples were sequenced with the pan-cancer panel,which covers the exons of 334 cancer-related genes and hotspot fusion introns.Performance results for these four diluted samples are summarized in Table 1 with allele frequency(AF)cutoffs of 0.1%,0.2%,and 0.5%.The SinoDuplex approach achieved a high sensitivity of 98.62% and specificity of 97.09%at an AF cut-off of 0.1%.Furthermore,a high correlation between the detected AF and expected AF was observed for all mutations in these four diluted samples(Figure 2).These performance assessment results demonstrated the ability of our approach to detect very low-frequency variants in plasma cfDNA samples.
Better utilization of raw sequencing data
In the process of generating SSCS consensus sequences,majority-based rule is often adopted to determine the consensus base at each position.At least three member reads in a SSCS family are needed to form a SSCS sequence in the original DS approach [10,21]. However, this majority-based approach is far from precise and many SSCS families with less three members are technically thrown away.In our Bayesianbased algorithm,we used Equation(2)to calculate the probability of each possible consensus base,selecting the base with maximum probability.Therefore,in our approach,even if there were only two reads in one SSCS family,we were still able to generate high-quality consensus sequences and thus more SSCS reads compared to the original approach.At the same time,we calculated the consensus base quality score using Equation(3).After the consensus sequences were generated,the mean base phred quality score increased from 38 to 80,indicating a significant reduction in the sequencing error rate(Figure S2).
In addition to reducing the cut-off of the SSCS family size from three to two,several modifications were implemented in the generation of DCS from two complementary SSCS families.If an SSCS family with only one read pair can form a DCS with another SSCS family,it is kept and thus more DCS consensus reads are produced.Moreover,if an SSCS family is missing its partner in a generated DCS consensus(singleton SSCS family),it is also kept when it has at least two members.To evaluate the performance of these changes,we compared our approach for all four diluted samples with the original duplex approach,which has cut-off of three for SSCS family size and only employs DCS sequences for variant calling(DCS_only),and an approach utilizing only SSCS sequences for variant calling(SSCS_only).Figure 3 shows the performance results of these three approaches for HD753M1 and HD753M2 at different AF cut-offs(detailed values are summarized in Table S5).In general,the SSCS_only approach had the best sensitivity but a low specificity due to its high mean depth.Unsurprisingly,SSCS_only generated more false positives due to PCR-amplified errors or DNA damage in single-strand sequences.In contrast,DCS_only had fewer false positives,as DCS is much more accurate.However,a large amount of data were disregarded in the DCS_only approach,as some low-frequency true mutations were classified as false negatives due to low depth.By employing both DCS and SSCS sequences for variant calling,our SinoDuplex approach achieved a better balance of sensitivity and specificity.
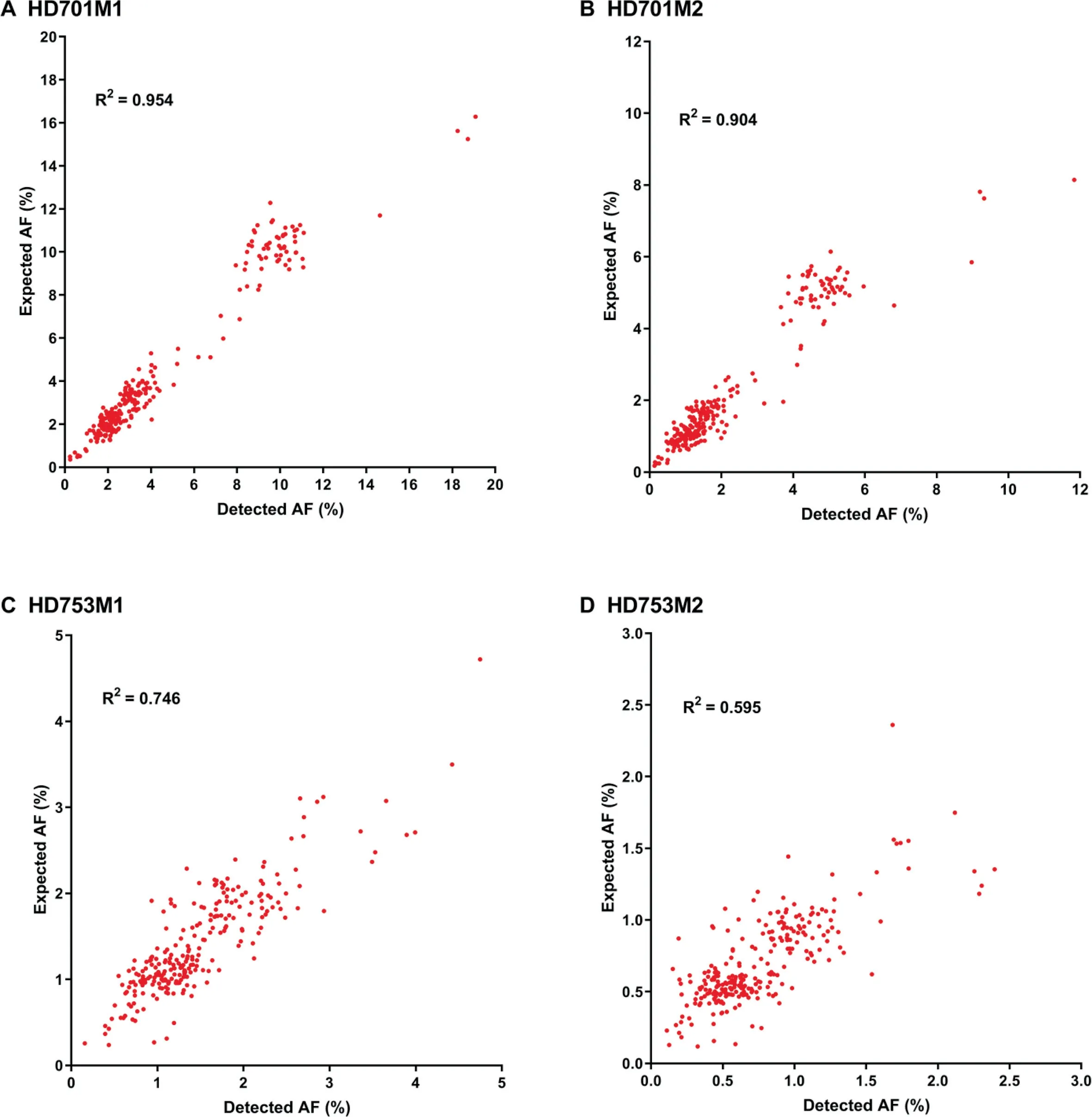
Figure 2 The correlation of detected AF with expected AF for mutations detected in four diluted samples
Impact of sequencing depth and SSCS family size
To determine the appropriate sequencing depth required for SinoDuplex to detect low-allele-frequency variant calling from cfDNA samples,we performed in silico downsampling for the sample HD753M2 at different depths from raw sequencing data and then calculated sensitivity and specificity after generating SSCS and DCS reads.Figure 4 shows that,as sequencing depth decreased,sensitivity dropped dramatically while specificity increased slightly at a limit of detection of 0.1%.The reason was that some rare mutations would not be detected at low depth and several other false positive variants appeared at higher sequencing depth.The best sensitivity and specificity results using SinoDuplex were achieved at a depth of 1831×.Obviously,there was an intriguing relationship between depth and family size.Therefore,we checked the distribution of SSCS family size with different sequencing depths.At a depth of 1968x,the family size of HD753M2 peaked expectably at 4,yielding high quality SSCS with optimal read numbers(Figure S3).However,a left shift of the peak generally occurred in the downsampling samples of lower depths.According to Equations(2)and(3),the SSCS family size itself also has an impact on the quality of consensus sequences.We counted the average base quality of the final consensus sequence and found a similar trend in peak family size.When increasing family size from 1 to 4, the corresponding base quality increased from 75 to 86.In contrast,lowering family size resulted in poorer base quality in the consensus sequences.In our experience,a reliable SSCS is generated when the read number in the family is about 3 to 6.More member reads in the same SSCS family would not contribute to the yield of SSCS but increase sequencing cost.
Validation results with ddPCR
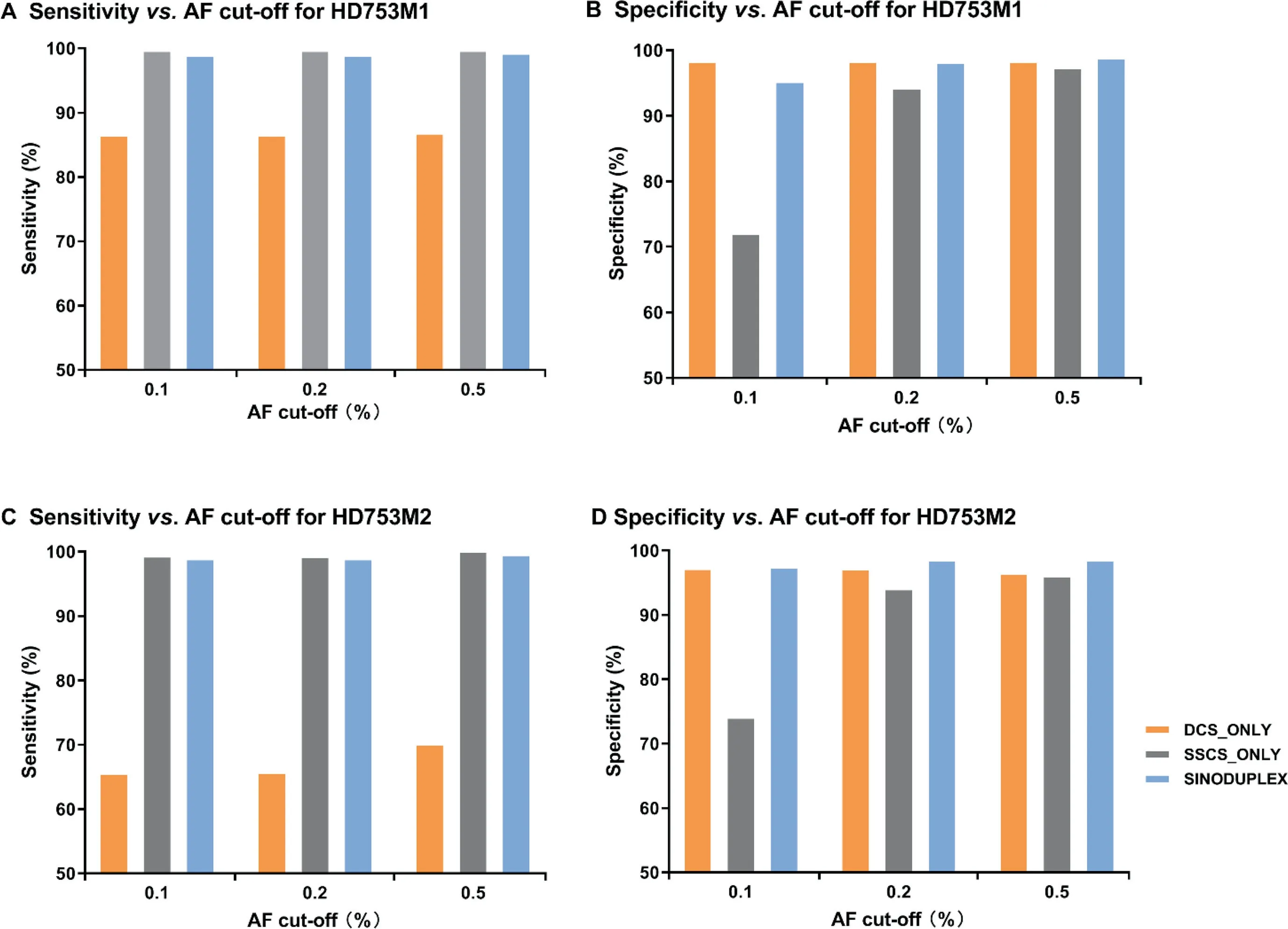
Figure 3 Comparison of the performance of three different approaches at different AF cut-offs
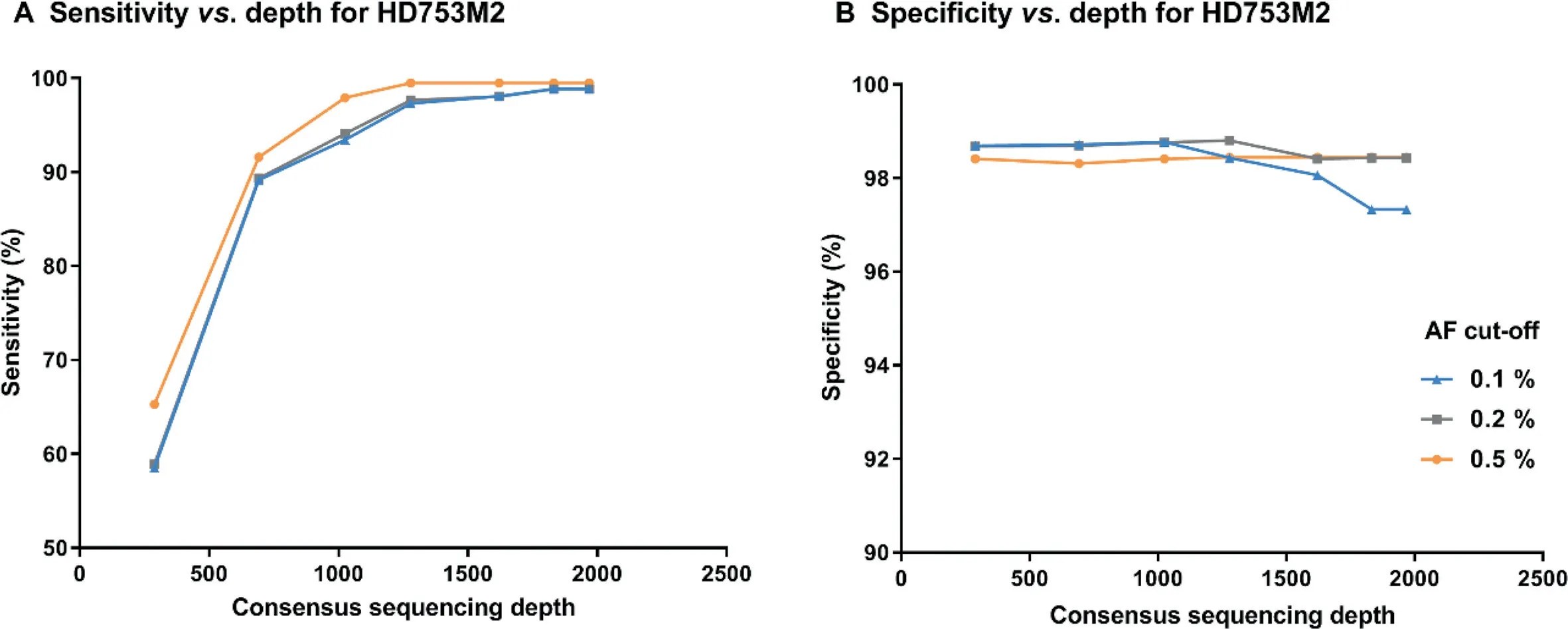
Figure 4 Impact of sequencing depth on the performance of SinoDuplex
Although good performance was observed in the reference standard samples,we also assessed the reliability of SinoDuplex in clinical samples and confirmed low-frequency mutations with ddPCR.Using customized panels of different sizes adapted for the SinoDuplex approach,a number of hotspot actionable mutations were detected with low frequency in cfDNA samples from lung cancer patients enrolled in our collaborating hospitals.Detection of variant allele frequencies below 5%remains a challenge and a subset of these variants detected by SinoDuplex with frequencies between 0.1%and 5%were validated by ddPCR.As summarized in Table 2,five of six actionable mutations from patient cfDNA samples detected by SinoDuplex were confirmed to have similar AFs by ddPCR.One mutation,EGFR T790M,had a frequency of 0.1%in patient P3 and was reported as negative with a very weak signal,as 0.1%is the limit of detection of ddPCR.Overall,our approach was consistent with ddPCR for these lowfrequency hotspot actionable variants.
Discussion
ctDNA refers to the fraction of cfDNA in a patient’s blood that originates from a tumor.Noninvasive access to cancerderived DNA is particularly attractive for solid tumors,allowing repeated sampling without invasive procedures.Advances in DNA sequencing technologies and our understanding of tumor molecular biology have resulted in increased interest in exploiting ctDNA as a tool to facilitate earlier detection of cancer and thereby improve therapeutic outcomes byenabling early intervention[38].However,the application of this method has been challenging due to the low sensitivity in analyzing trace amounts of ctDNA in blood. Many sequencing technologies and algorithms have been developed and optimized to improve the detection accuracy of low frequency variants from ctDNA samples.One of these methods,DS technology,is particularly sensitive and uses attachment of unique molecular identifiers (UMI) to DNA templates to detect and quantify low-frequency genetic alterations in ctDNA samples [10,21]. Its power comes from pooling together multiple descendants of both strands of the original DNA molecules,allowing true variants to be distinguished from PCR amplification and sequencing artifacts.However,the method’s reliance on multiple sequencing reads of the same molecule means that DS requires much larger sequencing capacity than conventional NGS to produce a given depth of sequencing data,making it prohibitively expensive for broad usage in clinical settings.Furthermore,every duplex experiment produces a substantial proportion of singleton SSCS families that cannot be used in the analysis and are technically thrown away.Despite the great promise of DS,methods for both the experimental and computational aspects of this technique are still evolving.
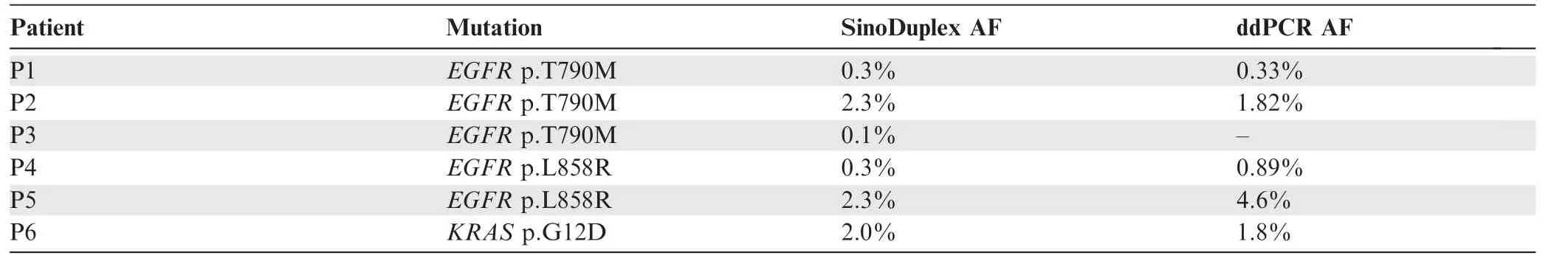
Table 2 Validation of hotspot actionable mutations detected by SinoDuplex with ddPCR assay
To process cfDNA DS data more efficiently,we developed an improved duplex barcoding strategy and a novel computational analysis algorithm called SinoDuplex to generate lowerror consensus sequences from raw sequencing data.Taking advantage of degenerate UMIs,our duplex adapters provide significant advantages over the 12 N duplex adapters originally described[10,21]and the commercially available 3 N duplex adapter(Integrated DNA Technologies,Coralville,IA).First,SinoDuplex adapters are much more cost-effective than commercially available 3 N duplex adapters and can be easily acquired by annealing each pre-defined UMI pair and pooling them together.In contrast,synthesis of 12 N duplex adapters requires a series of enzymatic and purification steps.Second,SinoDuplex adapters are sufficient to identify most original nucleic acid molecules possessing the same genomic coordinates(start and end positions)in a sample while using far fewer barcode combinations compared to 12 N duplex adapters.Finally,pre-defined UMIs with at least three edit distances ensure improved accuracy for identification, while degenerate UMIs may be affected by single-base mismatches introduced by amplification or sequencing errors,leading to erroneous identification.Figure S4 shows a detailed graphical depiction of these different duplex adapters.Moreover,compared to DS analysis approaches reported previously[10,20,21], two major improvements were implemented in SinoDuplex to generate consensus sequences more precisely and utilize raw sequencing data more efficiently.The first one is the adoption of Bayesian theory[Equations(1),(2),and(3)]to generate SSCS sequences and quality scores.The other one is a lowering of the read cut-off from three to two in generating SSC sequences and retaining singleton SSCS families in subsequent analyses.The consensus sequence generation step significantly reduced PCR amplification errors and sequencing errors.Retaining singleton SSCS consensus reads improved utilization of raw sequencing data and reduced sequencing costs.SinoDuplex can be easily integrated into any existing pipelines that analyze DS data.
Conclusions
In this study,we present SinoDuplex,a promising DS method aimed at improving the sensitivity of detection of lowfrequency mutations in cfDNA.Compared to original DS approaches, our new computational analysis algorithm increased the yield of DS data while making it more costeffective.The method significantly improves the sensitivity and specificity in detecting extremely low frequency variants in plasma cfDNA samples.The potential of our approach for routine clinical applications will be of great importance for physicians,providing them with a powerful tool to diagnose tumors,monitor tumor dynamics,and evaluate patient responses to targeted therapy.
Data availability
The source code of SinoDuplex is freely available for academic use from https://github.com/SinOncology/sinoduplex. The raw duplex sequencing data reported in this paper have been deposited in the Genome Sequence Archive [39] at the National Genomics Data Center,Beijing Institute of Genomics,Chinese Academy of Sciences/China National Center for Bioinformation (GSA: CRA001933), and are publicly accessible at https://bigd.big.ac.cn/gsa.
Authors’contributions
BH conceived the idea and supervised the study.BH and YR designed the algorithm.YR and DW implemented the algorithm and performed the main analysis.YZ performed the laboratory validation experiments.FL,YF,SX,LS,JL,and HD analyzed the results and contributed to the manuscript writing.YR,YZ,DW drafted the manuscript,and BH edited the manuscript. ALL authors read and approved the final manuscript.
Competing interests
The authors have declared that no competing interests exist.
Acknowledgments
This work was financed by Grant-in-aid for scientific research from the Guangzhou Science and Technology Plan projects of China(Grant No.201802020004).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2020.02.003.
ORCID
0000-0003-2132-252X(Ren Y)
0000-0002-0098-8974(Zhang Y)
0000-0003-2485-3019(Wang D)
0000-0003-1896-963X(Liu F)
0000-0001-5211-8930(Fu Y)
0000-0001-6910-7322(Xiang S)
0000-0002-5990-8457(Su L)
0000-0003-2148-9872(Li J)
0000-0001-8024-458X(Dai H)
0000-0002-4748-2882(Huang B)
 Genomics,Proteomics & Bioinformatics2020年1期
Genomics,Proteomics & Bioinformatics2020年1期
- Genomics,Proteomics & Bioinformatics的其它文章
- Acknowledgments to Reviewers 2019
- GPS 5.0:An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins
- MSIsensor-pro:Fast,Accurate,and Matchednormal-sample-free Detection of Microsatellite Instability
- Procleave:Predicting Protease-specific Substrate Cleavage Sites by Combining Sequence and Structural Information
- Epitranscriptomic 5-Methylcytosine Profile in PM2.5-induced Mouse Pulmonary Fibrosis
- CRISPR Screens Identify Essential Cell Growth Mediators in BRAF Inhibitor-resistant Melanoma