
Capsid protein from red-spotted grouper nervous necrosis virus induces incomplete autophagy by inactivating the HSP90ab1-AKT-MTOR pathway
2022-01-27 08:23:06WanWanZhangPengJiaXiaoBingLuXiaoQiChenJueHuaWengKunTongJiaMeiShengYi
Zoological Research 2022年1期
Wan-Wan Zhang, Peng Jia, Xiao-Bing Lu, Xiao-Qi Chen, Jue-Hua Weng, Kun-Tong Jia,*,Mei-Sheng Yi,*
1 School of Marine Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510000, China
2 Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai, Guangdong 519000, China
3 Guangdong Provincial Key Laboratory of Marine Resources and Coastal Engineering, Guangzhou, Guangdong 510000, China
4 Pearl River Estuary Marine Ecosystem Research Station, Ministry of Education, Zhuhai, Guangdong 519000, China
ABSTRACT As a highly important fish virus, nervous necrosis virus (NNV) has caused severe economic losses to the aquaculture industry worldwide.Autophagy, an evolutionarily conserved intracellular degradation process, is involved in the pathogenesis of several viruses.Although NNV can induce autophagy to facilitate infection in grouper fish spleen cells, how it initiates and mediates autophagy pathways during the initial stage of infection is still unclear.Here, we found that red-spotted grouper NNV (RGNNV)induced autophagosome formation in two fish cell lines at 1.5 and 3 h post infection, indicating that autophagy is activated upon entry of RGNNV.Moreover, autophagic detection showed that RGNNV entry induced incomplete autophagy by impairing the fusion of autophagosomes with lysosomes.Further investigation revealed that binding of the RGNNV capsid protein (CP) to the Lateolabrax japonicus heat shock protein HSP90ab1(LjHSP90ab1), a cell surface receptor of RGNNV,contributed to RGNNV invasion-induced autophagy.Finally, we found that CP blocked the interaction of L.japonicus protein kinase B (AKT) with LjHSP90ab1 by competitively binding the NM domain of LjHSP90ab1 to inhibit the AKT-mechanistic target of the rapamycin (MTOR)pathway.This study provides novel insight into the relationship between NNV receptors and autophagy,which may help clarify the pathogenesis of NNV.
Keywords: NNV; Autophagy; CP; HSP90ab1;AKT-MTOR pathway
INTRODUCTION
Nervous necrosis virus (NNV), a single-stranded RNA virus belonging to the Nodaviridae family, is a viral pathogen that infects more than 200 species of marine and freshwater cultured fish, including the orange-spotted grouper(Epinephelus coioides), turbot (Scophthalmus maximus), and sea bass (Lateolabrax japonicus) (Bandin & Souto, 2020).The massive mortality of NNV-infected individuals, especially thealmost 100% fatality rate in infected juveniles, has resulted in huge economic losses (Costa & Thompson, 2016).The NNV genome is split into two single-stranded RNA segments,encoding the viral polymerase and capsid protein (CP),respectively (Buonocore et al., 2019).As the only structural protein displayed on the NNV surface, CP is involved in the viral invasion and encapsidation processes.Recent research has indicated that CP determines host specificity and virulence of Betanodavirus (Iwamoto et al., 2004; Souto et al.,2015).However, the precise mechanism by which CP regulates the pathogenesis of NNV remains to be elucidated.
Autophagy is a highly conserved catabolic process that plays a critical role in maintaining intracellular homeostasis(Galluzzi et al., 2014).Various stress stimuli induce autophagy by inhibiting the mechanistic target of rapamycin (MTOR)signaling and/or activating AMP-activated protein kinase(AMPK) signaling (Cobbold, 2013; He & Klionsky, 2009).The process of autophagy begins with the formation of phagophores, which further form double membrane-delimited autophagosomes by elongating and enclosing the cytoplasmic constituent.The formation of autophagosomes involves the conversion of LC3 (Atg8) from its C-terminal free form (LC3-I)to its lipidated C-terminal state (LC3-II).These autophagosomes eventually fuse with lysosomes to form single-membrane autolysosomes, where degradation takes place (Boya et al., 2013; Yu et al., 2018).
Autophagy plays a vital role in the cellular response to pathogens but is a “double-edged sword” due to its complex relationship with pathogen infection (Levine & Klionsky, 2004;Shintani & Klionsky, 2004).On the one hand, autophagy functions as an intrinsic antiviral defense mechanism for the removal of intracellular viruses by lysosomal degradation(Liang et al., 1998; Tallóczy et al., 2006).On the other hand,some viruses can exploit host autophagy to facilitate their own replication, consequently causing host pathogenesis (Espert et al., 2007).In addition, an increasing number of viruses have developed complex strategies to regulate host autophagy at different stages of viral infection (Hu et al., 2015; Richetta et al., 2013; Yang et al., 2020).For example, the peste des petits ruminant virus (PPRV) can induce two successive waves of autophagy during the early and late infection stages,respectively.The first (early) wave of autophagy is induced during viral entry into cells and the second wave is induced during viral replication (Hu et al., 2015; Richetta et al., 2013;Yang et al., 2020).
Virus receptors on the host cell surface are vitally important for viral entry.Growing evidence suggests that some cell surface virus receptors have dual roles in facilitating viral entry and triggering autophagy (Joubert et al., 2009).For example,nectin4, a PPRV entry receptor, binds to the PPRV-H protein,leading to the induction of early wave autophagy (Hu et al.,2015; Yang et al., 2020).In addition, cell membrane surfacedistributed HSP90AA1, an avibirnavirus-binding receptor, can induce autophagy through the HSP90AA1-AKT-MTOR pathway in the early stage of infection (Wang et al., 2020).Research has also shown that NNV infection can induce autophagy in grouper fish spleen cells at 6–12 h post infection(hpi) (Li et al., 2020), although how autophagy is initiated by NNV invasion remains unclear.We previously reported that the heat shock protein 90ab1 (HSP90ab1) may be a universal red-spotted grouper NNV (RGNNV) entry receptor in various fish and facilitates RGNNV internalization through the clathrindependent endocytosis pathway (Zhang et al., 2020).In view of the important role of virus receptors in viral entry-induced autophagy, we investigated the effects of RGNNV entry on autophagy induction and clarified the role of L.japonicus HSP90ab1 (LjHSP90ab1) in RGNNV-induced autophagy at the early invasion stage.Furthermore, the underlying mechanism of LjHSP90ab1-mediated RGNNV entry-induced autophagy was explored.Our findings provide new insight into the relationship between autophagy and RGNNV and should contribute to the development of antiviral drugs.
MATERIALS AND METHODS
Cells, virus, and reagents
The L.japonicus brain cell line (LJB) was previously established from the brains of sea perch (Le et al., 2017) and cultured in Dulbecco‘s modified Eagle medium (DMEM)supplemented with 15% (v/v) heat-inactivated fetal bovine serum (FBS, Gibco, USA) at 28 °C.hMMES1 cells, derived from marine medaka embryo blastocysts, were cultivated at 28 °C in embryonic stem cell medium 4 (ESM4), as described previously (Yi et al., 2009, 2010).HEK293T cells were maintained in DMEM with 10% FBS and incubated at 37 °C with 5% CO2.
RGNNV was originally isolated from diseased sea perch larvae in Guangdong Province, China (Le et al., 2017).Viral stocks (1.0×108TCID50/mL) were proliferated in LJB cells and stored at ?80 °C for use.Ultraviolet (UV) inactivation of RGNNV was performed by UV light irradiation at room temperature (RT) for 1 h with a dose of 2 500 mJ (GS Gene Linker UV Chamber, Bio-Rad USA).Viral titer assay was performed to confirm that the UV-treated virus was successfully inactivated, as described previously (Zhang et al.,2019).
Mouse anti-Flag (Cat.No.M20008), anti-Myc (Cat.No.M20002), anti-His (Cat.No.M20001L), and rabbit anti-ACTB(Cat.No.P30002) antibodies were purchased from Abmart(China) and were used at a dilution of 1:4 000, respectively.Rabbit anti-LC3 (Cat.No.ab 128025), anti-SQSTM1 (Cat.No.ab 91526) and anti-human HSP90β (Cat.No.ab 236282)polyclonal antibodies were purchased from Abcam (USA) and were used at a dilution of 1:1 000, respectively.Rabbit anti-p-AKT (Ser473) (Cat.No.9271), anti-AKT (Cat.No.4691), antip-MTOR (Ser2448) (Cat.No.2971), anti-MTOR (Cat.No.2983), anti-mouse IgG horseradish peroxidase (HRP)-linked(Cat.No.7076), and anti-rabbit IgG HRP-linked antibodies(Cat.No.7074) were purchased from Cell Signaling Technology (USA) and were used at a dilution of 1:1 000,respectively.The 4‘,6-diamidino-2-phenylindole (DAPI) stain solution (Cat.No.D9542), insulin (Cat.No.I0305000), and phenylmethylsulfonyl fluoride (PMSF, Cat.No.P7626) were acquired from Sigma Aldrich (USA).The pmCherry-GFP-LC3 and LAMP1-RFP plasmids were purchased from REBIO(China) and MIAOLINGBIO (China), respectively.The specificity and sensitivity of anti-human HSP90β polyclonal antibodies to LjHSP90ab1 were assessed by western blotting(Supplementary Figure S1).
Plasmid construction
For expression plasmid construction, the open reading frame(ORF) cDNA of the RGNNV CP (GenBank accession No.:KP455642) was amplified by polymerase chain reaction (PCR)and cloned into the pCMV-Flag, pCMV-Myc, pDsRed2-C1,and pET-32a (+) vectors, which were acquired from Clontech(USA).The coding regions of LjHSP90ab1 were cloned into pCMV-Flag, pCMV-Myc, and pET-32a (+) vectors,respectively.The L.japonicus AKT gene was cloned into pCMV-Flag and pCMV-Myc vectors, respectively.The L.japonicus and marine medaka LC3 genes were cloned into the pEGFP-N3 vector, respectively.Serially truncated mutants of LjHSP90ab1 with Flag-tag, including pCMV-Flag-LjHSP90ab1-NC, pCMV-Flag-LjHSP90ab1-NM, pCMV-Flag-LjHSP90ab1-MC, pCMV-Flag-LjHSP90ab1-M, pCMV-Flag-LjHSP90ab1-C, and pCMV-Flag-LjHSP90ab1-N, were generated using the pCMV-Flag-LjHSP90ab1 plasmid as a template.Serially truncated mutants of CP with Flag-tag,including pCMV-Flag-CP-ΔARM, pCMV-Flag-CP-Δarm,pCMV-Flag-CP-ΔS, pCMV-Flag-CP-ΔLR, and pCMV-Flag-CPΔP plasmids, were generated using the pCMV-Flag-CP plasmid as a template.The PCR specific primers used are shown in Supplementary Table S1.The expression plasmids were all constructed using standard molecular biology techniques and examined via DNA sequencing (Hopp et al.,1988).
Viral infection
The LJB and hMMES1 cells were separately seeded into 6-well plates at 1×106cells per well and infected with RGNNV at a multiplicity of infection (MOI) of 3 or mock-infected with phosphate-buffered saline (PBS) for 1.5 and 3 h, respectively.The lysed cells were harvested for immunoblotting analysis.
Immunoblotting analysis
Immunoblot experiments were performed as described previously (Jia et al., 2013, 2015).The cell lysates were boiled for 10 min and separated by sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE), then transferred onto 0.22 μm polyvinylidene difluoride membranes(Millipore, USA), which were subsequently blocked with 5%nonfat dried milk in TBST buffer (25 mmol/L Tris-HCl,150 mmol/L NaCl, 0.1% Tween-20 (pH 7.5)) for 1 h at RT.Thereafter, the membranes were incubated with different primary antibodies at 4 °C overnight followed by HRP-conjugated secondary antibodies for 1 h at RT.Immunoreactive bands were detected with ECL immunoblotting detection reagents (Millipore, USA) after three additional washes with TBST buffer.Images were obtained with a Minichemi 420 chemiluminescence instrument (Sage Creation, China), and protein blot intensity was analyzed using Lane 1D software (v4.22) within the same linear range.
Transmission electron microscopy (TEM)
The LJB cells infected with RGNNV for 3 h were harvested and prepared for TEM, as described previously (Zhang et al.,2020).Cells were fixed with 2.5% glutaraldehyde (pH 7.4) and 2.0% osmium tetroxide in turn at 4 °C for 24 h.Ultrathin sections were observed under a Philips CM10 transmission electron microscope.
RNA interference
The designed short interfering RNAs (siRNAs) targeting LjHSP90ab1 were synthesized by the Ribobio Company(China) (Supplementary Table S2).The LJB cells were transfected with LjHSP90ab1 siRNA mixtures (100 nmol/L) or negative control (NC) (100 nmol/L) for 24 h according to the manufacturer‘s instructions, then infected with RGNNV(MOI=3) and harvested at 1.5 and 3 hpi for immunoblotting,respectively.
Protein treatments
The His-CP and His-HSP90ab1 fusion proteins were expressed in Escherichia coli BL21(DE3) with pET-32a (+)-CP and pET-32a (+)-LjHSP90ab1 plasmids, respectively, as described previously (Zhang et al., 2020).For protein stimulation, the LJB cells were precoated with His or His-CP proteins (500 ng/mL) for 1.5 and 3 h at 28 °C, respectively.The lysed cells were then harvested for immunoblotting analysis.To investigate the effects of the LjHSP90ab1 protein on RGNNV infection, RGNNV (MOI=3) was preincubated with His or His-LjHSP90ab1 proteins (500 ng/mL) at 4 °C for 3 h,then infected in LJB cells for 1.5 and 3 h at 28 °C,respectively.The lysed cells were then harvested for immunoblotting analysis.
Antibody stimulation
For antibody stimulation, the LJB cells were preincubated with anti-human HSP90β polyclonal antibody or IgG control (1:50)for 3 h at 4 °C, then infected with RGNNV (MOI=3) for 1.5 and 3 h, respectively.The lysed cells were then harvested for immunoblotting analysis.
Insulin treatment
The LJB cells were pretreated with insulin (1 μmol/L) for 6 h prior to RGNNV infection.Sterilized water was used for the control cells.At 3 h after RGNNV infection, cells were harvested and lysed with lysis buffer for autophagy detection by immunoblotting using appropriate antibodies.
Coimmunoprecipitation (Co-IP)
The Co-IP assays were performed as described previously(Jia et al., 2013).HEK293T cells in 25 cm2dishes were cotransfected with indicated plasmids using Lipofectamine 3000 (Invitrogen, USA).At 48 hpi, cells were washed twice with ice-cold PBS and lysed on ice with immunoprecipitation lysis buffer (Beyotime, China).For each sample, 500 μL of lysate was incubated with anti-Flag magnetic beads(MedChemExpress, USA) overnight at 4 °C with constant agitation.The beads were then washed five times with 1 mL of wash buffer.Finally, the immunoprecipitates and whole-cell lysates were resuspended in SDS loading buffer and subjected to immunoblotting analysis using indicated antibodies.
Transient transfection and confocal microscopy
The LJB, hMMES1, and HEK293T cells growing on glass coverslips in 12-well culture plates were transfected with indicated plasmids using Lipofectamine 3000 according to the manufacturer‘s protocols, as described previously (Zhang et al., 2020).After various treatments, cells were washed withPBS and fixed with 4% paraformaldehyde for 10 min.After washing three times, cell nuclei were stained with DAPI, then visualized and documented using a confocal microscope(LSM510; Zeiss, Germany).
Statistical analysis
All data were analyzed using SPSS v20.The statistical significance of differences between groups was determined using one-way analysis of variance (ANOVA).P<0.05 andP<0.01 indicated statistically significant and highly statistically significant, respectively.
RESULTS
RGNNV induces autophagosome accumulation in LJB and hMMES1 cells in early-stage infection
To explore whether RGNNV induces autophagy in the early stage of infection, we investigated the kinetics of autophagy in LJB and hMMES1 cells at 1.5 and 3 hpi.The LJB and hMMES1 cells were transfected withGFP-LC3plasmid for 24 h, then infected with RGNNV for 3 h.As shown in Figure 1,many small GFP-LC3 (an autophagosome marker) puncta were observed in the LJB (Figure 1A) and hMMES1 cells(Figure 1B) at 3 hpi, compared to the cytoplasmic distribution of GFP-LC3 in the mock-infected cells, thus indicating that RGNNV entry can induce autophagosome formation.Consistent with the GFP-LC3 fluorescence, double-membrane autophagosome-like vesicles were observed in the cytoplasm of RGNNV-infected LJB (Figure 1C) and hMMES1 cells(Figure 1D) at 3 hpi.To verify the finding that RGNNV entry induces autophagosome formation, the expression levels of LC3-I and LC3-II were analyzed by western blotting in the LJB and hMMES1 cells at 1.5 and 3 hpi.Compared with the mock-infected cells, endogenous LC3-II expression was strongly increased in the RGNNV-infected LJB (Figure 1E, F) and hMMES1 cells (Figure 1G, H) at both 1.5 and 3 hpi (P<0.05 andP<0.01, respectively).These results suggest that RGNNV invasion can trigger autophagosome accumulation in cultured cells.
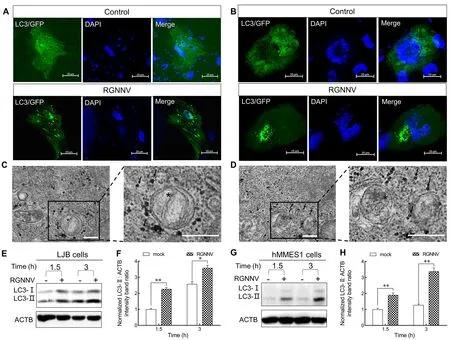
Figure 1 Characterization of RGNNV entry-triggered autophagosome accumulation
RGNNV invasion induces incomplete autophagy in LJB cells
To clarify the mechanism by which RGNNV entry triggers autophagosome accumulation, the protein levels of SQSTM1,a target digested through the autophagy-lysosome process(Klionsky et al., 2016), were analyzed in RGNNV-infected LJB cells.No change in SQSTM1 levels was observed in the RGNNV-infected LJB cells at 1.5 and 3 hpi (Figure 2A, B).Similar results were also observed in the RGNNV-infected hMMES1 cells (Figure 2C, D), indicating that RGNNV prevents autophagosomal degradation.To determine whether RGNNV blocks the fusion of autophagosomes with lysosomes,mCherry-GFP-LC3orGFP-LC3andRFP-LAMP1plasmids were transfected into LJB cells, respectively.As shown in Figure 2E, many yellow autophagosomes were detected in the RGNNV-infected cells transfected with themCherry-GFP-LC3plasmid in comparison to the mockinfected LJB cells at 1.5 and 3 hpi.In addition, no colocalization between LC3 and LAMP1 (a lysosome marker)was observed in the RGNNV-infected LJB cells cotransfected withGFP-LC3andRFP-LAMP1plasmids at 1.5 and 3 hpi(Figure 2F), thus indicating that the autophagosomes did not fuse with the lysosomes.Thus, RGNNV appears to induce incomplete autophagy by impairing the fusion of autophagosomes with lysosomes in LJB cells at the early stage of infection.
CP is critical for RGNNV-induced autophagy in earlystage infection
Previous studies have indicated that RGNNV CP can induce autophagy in grouper spleen cells (Li et al., 2020).To investigate whether CP also triggers autophagy in LJB cells,pDsRed2-C1-CPandGFP-LC3plasmids were cotransfected into LJB cells for 12 and 48 h.Confocal microscopy revealed many LC3 ring-like structures and small puncta in the CP-transfected cells but not in the control cells (Figure 3A),indicating that CP can trigger autophagy in the LJB cells.To further identify the CP domain required for autophagy induction, the LJB cells were transfected with various CP mutants (Supplementary Figure S2).As shown in Figure 3B and 3C,pCMV-Flag-CP-ΔSandpCMV-Flag-CP-ΔPsignificantly up-regulated the level of LC3-II, similar to the fulllength CP, whereas thepCMV-Flag-CP-ΔARM,pCMV-Flag-CP-Δarm, andpCMV-Flag-CP-ΔLRmutants lost their stimulatory abilities.These findings were further supported by confocal microscopy, which showed that the small GFP-LC3 ring-like structures and puncta were only observed in thepCMV-Flag-CP-ΔS-,pCMV-Flag-CP-ΔP-, andpCMV-Flag-CP-transfected LJB cells (Figure 3D).Thus, these results suggest that the ARM, arm, and LR domains of CP are sufficient to induce autophagy.
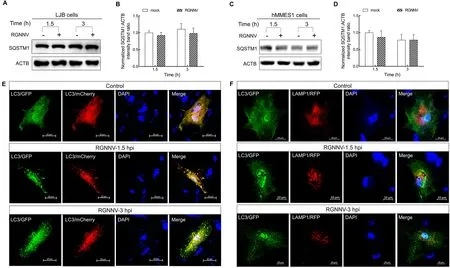
Figure 2 RGNNV induced incomplete autophagy
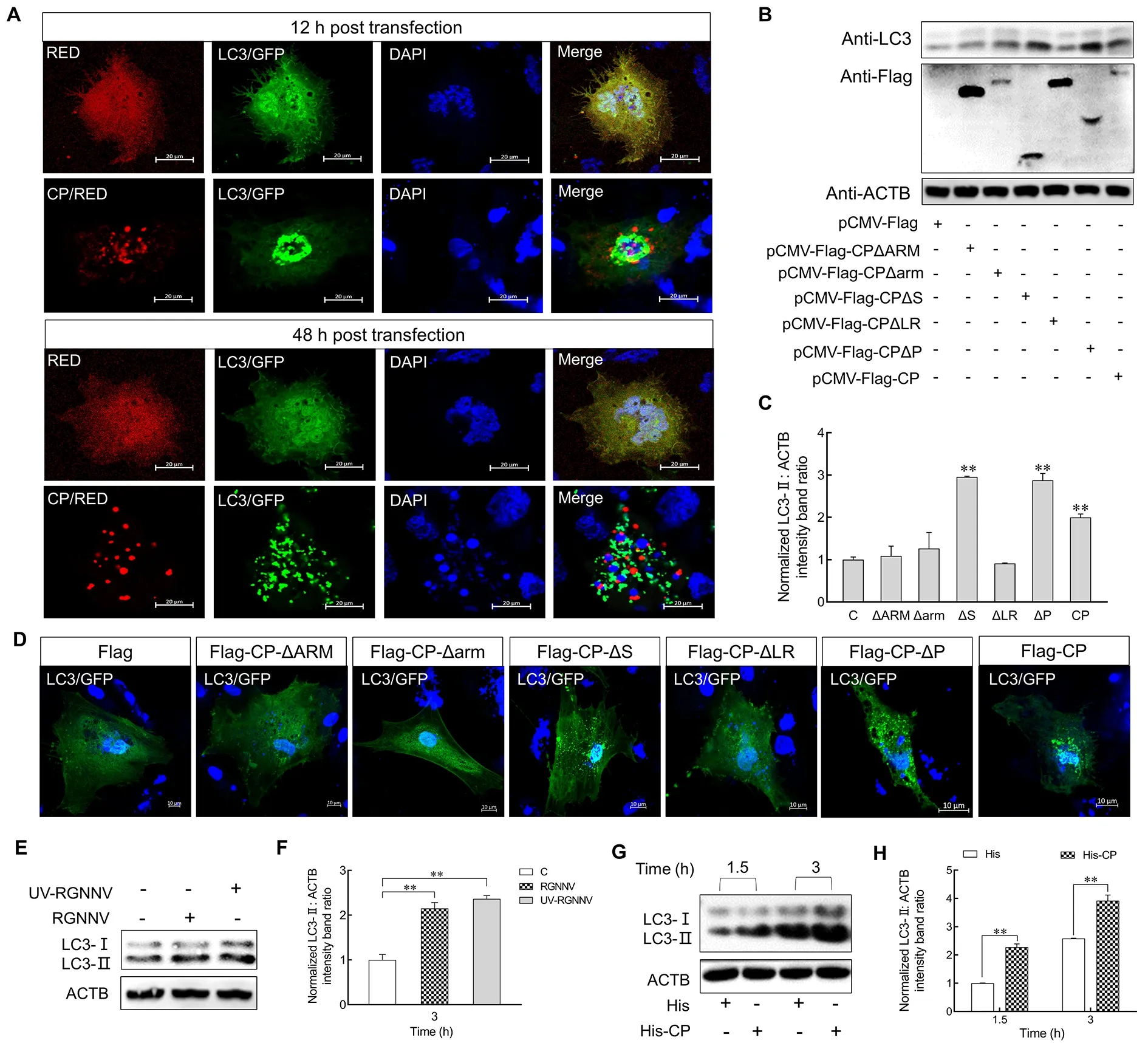
Figure 3 CP was sufficient for RGNNV to induce autophagy
To determine whether CP is responsible for the incomplete autophagy induced by RGNNV invasion, we detected LC3-II expression in the LJB cells post RGNNV and UV-irradiated RGNNV (UV-RGNNV) treatment.Results showed that the expression level of LC3-II was up-regulated in the RGNNV-and UV-RGNNV-infected LJB cells compared to the mock group at 3 hpi (P<0.01) (Figure 3E, F).LJB cells were also incubated with purified His or His-CP fusion proteins for 1.5 and 3 h, with the level of endogenous LC3-II then examined.As shown in Figure 3G, H, LC3-II was up-regulated in His-CP protein-treated cells compared with the His protein treatment group at 1.5 and 3 h (P<0.01).These results indicate that CP is sufficient to induce autophagy at the early stage of RGNNV infection.
LjHSP90ab1 contributes to RGNNV-induced autophagy
We previously reported that membrane-distributed HSP90ab1,as a receptor of RGNNV, can interact with CP to promote RGNNV entry (Zhang et al., 2020).Thus, we examined whether LjHSP90ab1 participates in RGNNV-induced autophagy in the early stage of infection.As shown in Figure 4, knockdown of LjHSP90ab1 by siRNA significantly inhibited RGNNV-induced autophagy at 1.5 and 3 hpi(Figure 4A, B), whereas overexpression of LjHSP90ab1 in LJB cells potentiated autophagy (Figure 4C, D).Likewise, the preincubation of recombinant LjHSP90ab1 protein (His-LjHSP90ab1) with RGNNV significantly disrupted induction of autophagy at 1.5 and 3 hpi (Figure 4E, F).Additionally, LC3-II expression was highly up-regulated in anti-HSP90β antibodytreated LJB cells with or without RGNNV infection in comparison with the immunoglobulin G (IgG)-treated cells(Figure 4G, H).These findings imply that LjHSP90ab1 is involved in autophagy induction in the early entry stage of RGNNV infection.
RGNNV entry induces autophagy through AKT-MTOR pathway
The AKT-MTOR pathway is reported to be a negative regulator of autophagy induction (Liang et al., 2016).To investigate whether RGNNV entry impacts the AKT-MTOR pathway to induce autophagy in LJB cells, we examined AKT and MTOR activity in LJB cells at 1.5 and 3 hpi.As shown in Figure 5, compared with the mock-infected cells, the level of LC3-II increased markedly in the RGNNV-infected cells at 1.5 and 3 hpi (Figure 5A, B).Simultaneously, the levels of phosphorylated AKT (p-AKT) and MTOR (p-MTOR) were down-regulated during RGNNV entry in the LJB cells(Figure 5A, C, D).These results indicate that autophagy triggered by RGNNV entry is associated with the AKT-MTOR pathway.To further validate the role of AKT and MTOR in RGNNV-induced autophagy, insulin was used to activate the AKT-MTOR pathway before RGNNV infection.As expected,the levels of p-AKT and p-MTOR increased markedly in the RGNNV-infected LJB cells with insulin treatment compared to those without insulin treatment (Figure 5E–H).In addition, the level of LC3-II decreased in the RGNNV-infected LJB cells treated with insulin in comparison to those not treated with insulin (Figure 5F).These findings indicate that RGNNV entry induces autophagy in LJB cells by inhibiting the AKT-MTOR pathway.
CP and LjHSP90ab1 mediate RGNNV entry-induced autophagy via AKT-MTOR pathway
To examine whether CP and LjHSP90ab1 contribute to RGNNV entry-induced autophagy via the AKT-MTOR pathway, we treated LJB cells with His-CP or His proteins and subsequently examined the levels of p-AKT and p-MTOR.As shown in Figure 6A–C, p-AKT and p-MTOR decreased significantly at 1.5 and 3 hpi in the His-CP-treated cells compared with the His control, indicating that CP triggered autophagy by suppressing activation of the AKT-MTOR pathway.Furthermore, LJB cells were treated with IgG or anti-HSP90β antibodies, followed by RGNNV infection or not.As shown in Figure 6D–F, levels of p-AKT and p-MTOR were significantly down-regulated in the anti-HSP90β antibodytreated cells in comparison to the IgG-treated cells without RGNNV infection.After RGNNV infection, p-AKT and p-MTOR were persistently down-regulated in the anti-HSP90β antibody-treated LJB cells, suggesting that anti-HSP90β antibodies inhibited the phosphorylation of AKT and MTOR in LJB cells.Additionally, RGNNV was incubated with His or His-HSP90ab1 proteins for 3 h, respectively, with the LJB cells then infected with RGNNV for 1.5 and 3 h.Results showedthat the expression levels of p-AKT and p-MTOR increased in the LJB cells treated with His-LjHSP90ab1-incubated RGNNV compared to the control (Figure 6G–I), suggesting that the recombinant LjHSP90ab1 proteins counteract the effects of RGNNV inhibiting phosphorylation of MTOR and AKT.
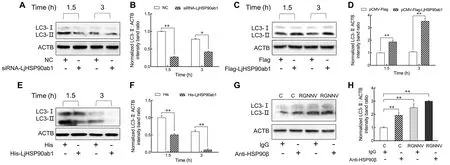
Figure 4 LjHSP90ab1 was responsible for RGNNV entry-induced autophagy
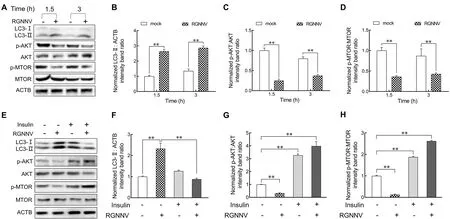
Figure 5 RGNNV induced autophagy at early infection stage by inhibiting AKT-MTOR pathway
Our previous study indicated that marine medaka HSP90ab1 overexpression can independently facilitate the internalization of RGNNV in RGNNV-impenetrable HEK293T cells (Zhang et al., 2020).To confirm whether LjHSP90ab1 can mediate RGNNV entry-induced autophagy,pCMV-Flag-LjHSP90ab1orpCMV-Flagplasmids were transfected into HEK293T cells, respectively, followed by RGNNV infection.As shown in Figure 6J–M, LjHSP90ab1 overexpression increased the level of LC3-II but reduced the levels of p-AKT and p-MTOR in the RGNNV-infected HEK293T cells compared with that in the RGNNV-infected cells without LjHSP90ab1 transfection.In addition,mCherry-GFP-LC3andpCMV-Flag-LjHSP90ab1orpCMV-Flagplasmids were transfected into HEK293T cells, respectively, followed by RGNNV infection.Compared to the HEK293T cells without LjHSP90ab1 transfection, many yellow puncta were observed in the LjHSP90ab1-overexpressing HEK293T cells (Figure 6N).These data indicate that RGNNV induces autophagy via the AKT-MTOR pathway by interacting with LjHSP90ab1.
CP attenuates interactions between LjHSP90ab1 and AKT
HSP90 is reported to regulate AKT kinase activity by its interactions with AKT (Sato et al., 2000).Given that LjHSP90ab1 interacts with CP, we further investigated the relationship between CP, LjHSP90ab1, and LjAKT in regulating RGNNV-induced autophagy.As seen in Figure 7A,the Co-IP assays showed that LjHSP90ab1 co-precipitated with LjAKT.To determine the LjHSP90ab1 domain required for its interactions with LjAKT,pCMV-Myc-LjAKTand different LjHSP90ab1 domain recombinant plasmids (Figure 7B) were cotransfected into HEK293T cells, respectively, followed by Co-IP assays.As shown in Figure 7C, the LjHSP90ab1-NM domain was associated with LjAKT, whereas the other LjHSP90ab1 domains lost their abilities to interact with LjAKT,indicating that the NM domain of LjHSP90ab1 is crucial for its interactions with LjAKT.Moreover, the HEK293T cells were cotransfected withpCMV-Myc-LjHSP90ab1andpCMV-FlagorpCMV-Flag-LjAKTfor 48 h, followed by treatment with His or His-CP proteins for 3 h.As shown in Figure 7D, the interactions between LjAKT and LjHSP90ab1 were weaken in the His-CP-treated cells compared to the His-treated cells,indicating that CP blocked the association between LjAKT and LjHSP90ab1.Collectively, these results suggest that CP competes with LjAKT to bind to LjHSP90ab1.
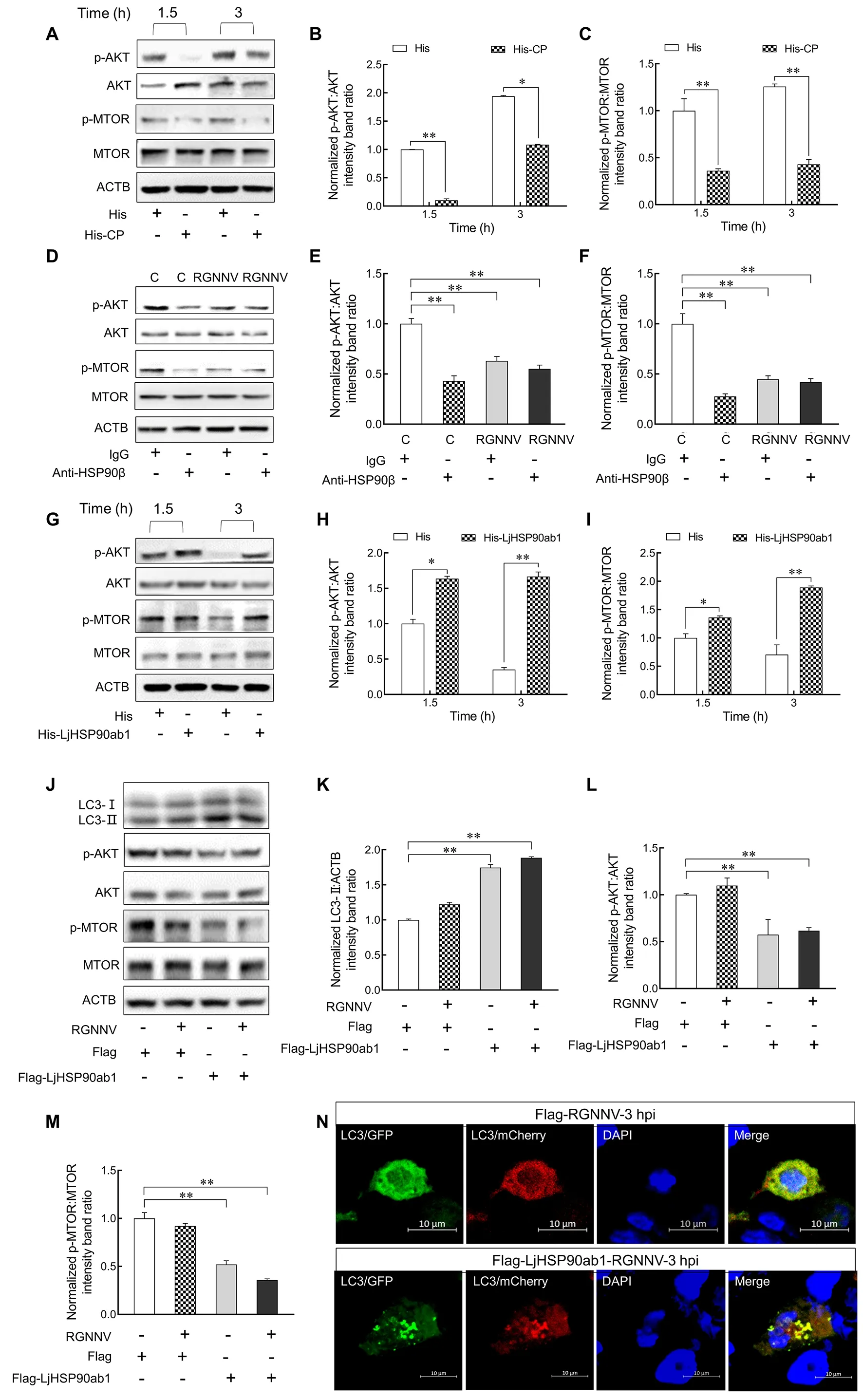
Figure 6 CP and LjHSP90ab1 mediated RGNNV entry-induced autophagy via AKT-MTOR pathway
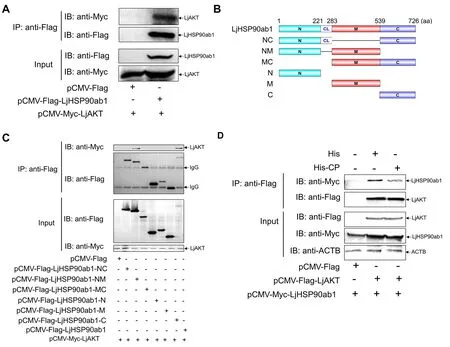
Figure 7 CP attenuated interactions between LjHSP90ab1 and AKT
DISCUSSION
Virus infection is associated with the autophagic process and viruses have developed diverse strategies to trigger autophagy during different stages of their life cycle, such as entry, replication, assembly, and release.For example,autophagy is induced during human papillomaviruses type 11 entry in human keratinocytes (Han et al., 2020).Porcine epidemic diarrhea virus replication but not viral entry triggers autophagy (Lin et al., 2020a).RGNNV induces autophagy at the viral replication stage in grouper spleen cells (Li et al.,2020).However, little is known about how autophagy occurs in the early stage of RGNNV infection.In the current study, we found that RGNNV invasion induced autophagy in both LJBand hMMES1 cells, indicating the autophagy induced by RGNNV invasion is independent of viral replication and is not cell-type specific.Furthermore, we found that RGNNV invasion blocked autophagolysosomal fusion, suggesting that incomplete autophagy is induced during RGNNV entry in LJB cells.Increasing evidence has demonstrated that viruses can modulate the autophagic mechanism to escape degradation.For example, upon entry into host cells, certain viruses induce incomplete autophagy by enhancing lysosomal acidification or blocking the fusion of autophagosomes with lysosomes to escape lysosomal degradation (Liao et al., 2020; Lin et al.,2020b).Thus, we propose that RGNNV may employ a similar strategy to prevent autophagic degradation and promote viral replication in infected cells.However, further studies are needed to clarify the precise mechanisms.
Various viruses have developed diverse strategies forregulating autophagy by encoding certain proteins, such as matrix protein 2 of influenza A virus (IAV) (Gannagé et al.,2009), Nef of HIV (Kyei et al., 2009), and ICP34.5 of herpes simplex virus 1 (HSV-1) (Orvedahl et al., 2007).HPV type 3 infection induces autophagy via its matrix protein to suppresses interferon responses (Ding et al., 2017).In African swine fever virus infection, the E199L protein triggers complete autophagy in HEK293T and Vero cells (Chen et al.,2021).In this study, we found that CP triggered autophagy in LJB cells, similar to that observed in grouper spleen cells (Li et al., 2020), indicating that CP-induced autophagy is universal in different fish cells.Previous study has indicated that the CP ARM domain is important for virus particle assembly (Chen et al., 2015).In earlier research, we showed that the LR domain is a critical region for HSP90ab1 interactions (Zhang et al.,2020).At present, however, the function of arm remains unknown.Here, our results showed that the CP ARM, arm,and LR domains were necessary for CP to induce autophagy,suggesting that these domains may have novel molecular functions during NNV infection.In addition, we found that UVRGNNV treatment triggered autophagy in LJB cells, indicating the dependence of CP in inducing autophagy during RGNNV invasion and the existence of other autophagy mediators that interact with CP on the cell surface.
Mounting evidence suggests that autophagy induced by pathogen invasion is dependent on pathogen receptors (Viret et al., 2018).The highly conserved 90 kDa heat shock protein(HSP90) plays crucial roles in the virus life cycle.Several studies have demonstrated that viral proteins can bind to HSP90 to mediate the autophagy pathway (Bekki et al., 2015;Mori et al., 2015).For example, IAV infection induces autophagy by the viral hemagglutinin glycoprotein binding to its HSP90AA1 receptor on the cell surface (Wang et al.,2020).Upon classical swine fever virus (CSFV) infection,increased interactions between the CSFV protein NS5A-3 and HSP90AB1 leads to autophagy activation through the MTOR signaling pathway (Xie et al., 2021).Our earlier study demonstrated that HSP90ab1 is a cellular receptor of RGNNV and LjHSP90ab1 interacts with CP (Zhang et al., 2020).Thus,we speculated that LjHSP90ab1 may be involved in RGNNV entry-induced autophagy.To verify this hypothesis, we carried out a series of experiments.Firstly, we found that overexpression of LjHSP90ab1 strengthened the formation of RGNNV-induced autophagosomes, whereas LjHSP90ab1 knockdown had the opposite effect.Likewise, RGNNV-induced autophagy was enhanced by blocking LjHSP90ab1 with anti-HSP90β antibodies but inhibited by pretreating the virus with recombinant LjHSP90ab1 protein.Notably, RGNNV triggered autophagy in the LjHSP90ab1-overexpressing HEK293T cells.These results suggest that LjHSP90ab1 is sufficient for RGNNV to initiate autophagy upon viral invasion.Thus, LjHSP90ab1 may exert dual roles, promoting RGNNV entry and inducing autophagy in the early stage of infection.
The regulation of autophagy involves diverse signaling cascades, including serine/threonine kinase MTOR, which is well known in mammalian cells for its participation in autophagy (Li et al., 2015).MTOR can integrate with upstream signals, such as PI3K/AKT and AMPK, to transduce signals for the initiation of autophagy.Previous studies have reported that coxsackievirus B3 infection inhibits the PI3K-AKT-MTOR signaling pathway to activate autophagy (Chang et al., 2017).Zika virus induces autophagy in host cells via the AKT-MTOR signaling pathway (Liang et al., 2016).In foot-and-mouth disease virus (FMDV)-infected cells, the phosphorylation of AKT-S473 and MTOR-S2448 is inhibited, resulting in inactivation of the AKT-MTOR signaling pathway, leading to autophagy (Sun et al., 2018a).HSP90 maintains AKT kinase activity through interactions with AKT, and inhibition of AKT binding to HSP90 results in AKT dephosphorylation and decreased AKT kinase activity (Sato et al., 2000).Accumulating evidence implicates the involvement of HSP90 in the AKT-MTOR signaling pathway and in virus-induced autophagy.For example, knockdown of HSP90 in human lung cells by siRNA decreases the level of p-AKT (Sun et al.,2018b).Increased interactions between HSP90AA1 and the IAV protein PB2 modulate the AKT-MTOR signaling pathway,leading to IAV-induced autophagy and enhanced viral RNA synthesis (Wang et al., 2020).Considering the relationship between HSP90 and the AKT-MTOR signaling pathway, we explored whether RGNNV entry induced autophagy through the LjHSP90ab1-AKT-MTOR axis.Our results showed that RGNNV entry induced autophagy via inactivation of the AKTMTOR signaling pathway.
AKT is an important client protein of HSP90 (Wang et al.,2019).Certain viruses trigger autophagy by disturbing HSP90 and AKT interactions.For instance, the infectious bursal disease virus (IBDV)-encoded VP2 protein can bind to HSP90AA1, leading to dissociation of p-AKT from HSP90AA1 and the dephosphorylation of MTOR and activated autophagosome formation (Hu et al., 2015).HA1 of IAV suppresses the AKT/MTOR pathway by disturbing the interactions between HSP90AA1 and AKT (Wang et al.,2020).In the present study, we found that LjHSP90ab1 regulated the level of p-AKT and interacted with LjAKT.Furthermore, the NM domain of LjHSP90ab1 was responsible for its interaction with LjAKT.In an earlier study, we found that the NM domain of HSP90ab1 is also required for its interactions with CP.Thus, we speculated that CP may compete with LjAKT for binding to LjHSP90ab1.This hypothesis was confirmed in the current study, whereby CP weakened the interactions between LjAKT and LjHSP90ab1.Thus, CP suppressed the AKT/MTOR pathway by inhibiting the interactions between HSP90ab1 and AKT.Previous research has reported that the eIF2α and MTOR proteins are involved in RGNNV replication-induced autophagy (Li et al.,2020).Considering that RGNNV entry and replication can induce autophagy, whether RGNNV induces two waves of autophagy during infection through distinct pathways needs to be further studied.
Our study showed that RGNNV invasion induced incomplete autophagy via its CP.Further mechanistic study showed that CP competed with LjAKT to bind to LjHSP90ab1,which, in turn, induced autophagy by inactivating the AKTMTOR signaling pathway (Figure 8).Our findings revealed the molecular mechanism underlying RGNNV invasion-induced autophagy, thus providing new insights into the pathogenesis of RGNNV and the development of effective antiviral strategies.
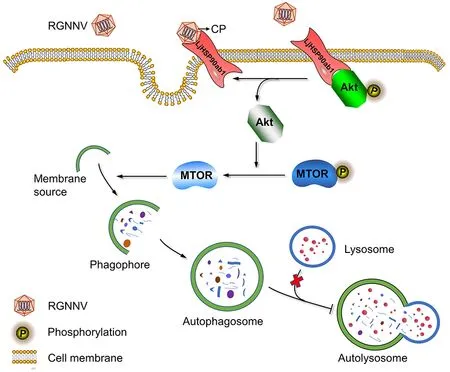
Figure 8 Proposed model for RGNNV entry-induced autophagy by blocking HSP90ab1/AKT/MTOR axis in LJB cells
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
All authors contributed to the study conception and design.K.T.J., M.S.Y., and W.W.Z.designed the research.W.W.Z.,P.J., X.B.L., X.Q.C., and J.H.W.conducted the research.W.W.Z., P.J., and X.B.L.provided acquisition, analysis, and interpretation of the data, and statistical analysis.W.W.Z.,K.T.J., and P.J.wrote the first draft of the manuscript.M.S.Y.and K.T.J.revised the manuscript.All authors read and approved the final version of the manuscript.

- Zoological Research的其它文章
- Rapid genetic divergence and mitonuclear discordance in the Taliang knobby newt (Liangshantriton taliangensis, Salamandridae, Caudata) and their driving forces
- Lack of transcriptional coordination between mitochondrial and nuclear oxidative phosphorylation genes in the presence of two divergent mitochondrial genomes
- Sichuan hot-spring snakes imperiled: reason, situation,and protection
- A new species of the genus Kurixalus (Anura:Rhacophoridae) from Sichuan Province, southwestern China
- A new species of Occidozyga Kuhl and van Hasselt,1822 (Anura: Dicroglossidae) from Southern Guangxi, China
- Role of Cyp19a1 in the female pathway of a freshwater turtle species (Mauremys reevesii) with temperaturedependent sex determination