
Promoting wetting of Mg on the SiC surfaces by addition of Al,Zn and Zr elements:A study via first-principl calculations
2022-07-26 11:41:36WeibingGuoWenshnBinHitoXueXiomingZhng
Journal of Magnesium and Alloys 2022年6期
Weibing Guo ,Wenshn Bin ,Hito Xue ,Xioming Zhng
aSchool of Materials Science and Engineering,Hebei University of Technology,Tianjin 300401,China
b State Key Laboratory of Advanced Welding and Joining,Harbin Institute of Technology,Harbin 150001,China
Abstract When preparing SiC/Mg composites,alloy elements play key roles in the nucleation and interfacial wetting.In this paper,effects of Al,Zn and Zr additions on the Mg/SiC interfacial bonding properties were investigated comprehensively via method of first-principl calculations.Mg(0001)/SiC(0001) interfaces with different terminations and stacking sequences were built and C Ⅱ-T C Ⅱtop interface has the largest work of adhesion (Wad).Zn dopants can not improve the Wad for both C-T and Si-T interfaces.Al atom can only strengthen the C-T interface.Zr addition can greatly improve the Wad for both C-T and Si-T interfaces.Wad of C-T interfaces can reach up to 11.55 J/m2 and 12.55 J/m2 after doping 1 monolayer (ML) of Al and Zr atoms.Larger Wad can lead to lower contact angles of Mg on SiC surfaces,which can improve wetting and nucleation in SiC/Mg composites.Analysis of electronic structure shows that Al-C and Zr-C bonds have more covalent composition than the Mg-C bond,which is responsible for the improvement of interfacial bonding strength.Experimental results in references were also analyzed,which are in well agreement with our calculation results.
Keywords: Mg alloy;SiC;Wetting;Electronic structure;First-principle calculations.
1.Introduction
Mg alloys have excellent properties of high specifi strength and low density,which make them as potential candidates in automotive,electronic,and aerospace industries[1,2].Recently,Mg and its alloys are also found to be potential materials for energy storages [3,4] due to high gravimetric and volumetric storage capacities.However,Mg alloys also have shortcomings of low strength,poor room temperature toughness and ductility,which have limited the wide applications of magnesium.In conventional processes,alloy elements of Al,Zn,Y,Zr and Ga are always added into Mg alloys to improve their mechanical properties [5].In order to improve specifi strength,creep resistance,thermal shock resistance and corrosion resistance further,some ceramic particles (such as SiC [6],TiC [7],ZrB2[8],Al2O3[9]) are added when preparing Mg matrix composites [10].Among these ceramics,SiC is common used for its properties of high strength,elastic modulus and hardness [11].SiC/pure Mg [12],SiC/AZ80[13],SiC/AZ61 [14,15] SiC/AZ91 [16,17] and SiCw/ZK60[18] composites were prepared by different processes.
A challenge to prepare SiC/Mg composites is the poor wettability between Mg melt and SiC ceramics.Additions of alloy elements to the SiC/Mg interfaces play key role in wetting and nucleation of the composites,which will influenc the microstructure and performance of composites further.First-principle calculation is based on density functional theory.Results of work of adhesion [19] and electronic structures [20] can be obtained through first-principl calculations,which can help us to understand the interface from atomic or electronic level.Adhesion properties and electronic structures of Mg/TiB2[21],Mg/ZrB2[22],Mg/Al4C3[23] and Mg/Al2CO [24] interfaces were all studied through first-principl calculations.
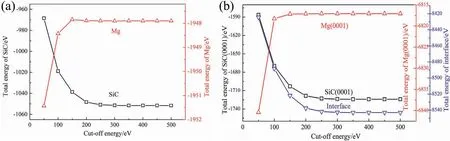
Fig.1.The relationships between cut-off energy and total energies of (a) SiC and Mg crystals,(b) surface and interface structures.
However,little study focused on the influenc mechanism of alloy elements on the SiC/Mg interface,which makes it difficul to design the composition of Mg alloy matrixs.Al[25,26],Zn [27,28] and Zr [29] are the common used alloy elements in Mg alloys and composites.In this work,through method of first-principl calculations,we examine that whether Al,Zn and Zr alloy elements can promote interfacial wetting between Mg alloy and SiC particles.The interfacial bonding properties and electronic structures are analyzed and discussed.The results can help us to optimize alloy compositions for SiC/Mg composites.
2.Calculation methods
Code of CASTEP (Cambridge Serial Total Energy Package),which is in framework of DFT (density functional theory),was used to study the energies and properties of the interfaces [30].The generalized gradient approximation (GGA)in Perdew-BurkeErnzerhof (PBE) was used as the exchangecorrelation functional.Method of ultrasoft pseudopotential was used to describe the interactions between ionic core and valence electrons.The valence electronic configuration were 2p63s2for Mg,2s23p2for C,3s23p2for Si,3s23p1for Al,4s24p64d25s2for Zr,and 3d104s2for Zn,respectively.Selection of cut-off energy has influenc on calculation accuracy and computing time.The relationship among cut-off energy and total energies of crystals (Mg and SiC),surfaces (Mg(0001) and SiC(0001)) and interface (Mg/SiC)structures are shown in Fig.1.When the cut-off energy is larger than 400 eV,energies of different structures all change little.Therefore,in the following calculation process,the cutoff energies of the plane waves were all set to be 400 eV.The Brillouin zone was sampled with a Monkhorst-Packk-point mesh.Thek-points meshing were 6×6×6 for the bulks and 6×6×1 for the interface and surface slabs.The tolerance was 1.0×10?5eV/atom when calculating energy.In the relaxation process,the convergence tolerances of stress,displacement and force were 0.05 GPa,0.001 °A and 0.03 eV/°A,respectively.All the calculation parameters were selected based on convergence test to balance the calculation accuracy and computing time.
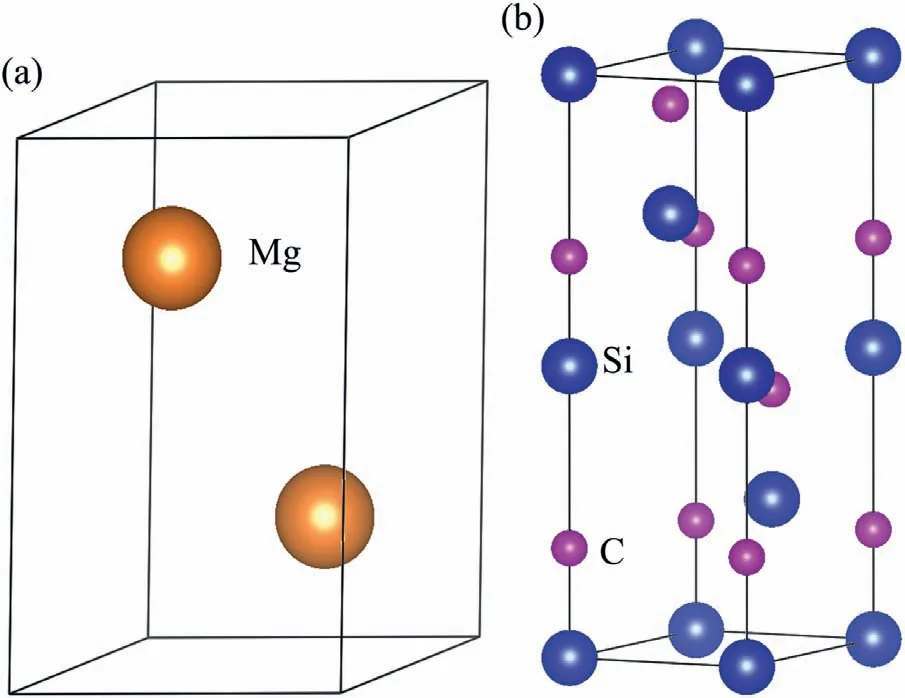
Fig.2.Crystal structures (a) Mg,(b) 4H-SiC (0001).
3.Results and discussion
3.1.Bulk properties of Mg and 4H-SiC
Fig.2 shows the structures of Mg and 4H-SiC crystals.Mg crystal has HCP structure (P63/MMC).The lattice constants of Mg are:a=b=3.2094 °A,c=5.2105 °A andα=β=90°,γ=120°.The 4H-SiC crystal has space group of P63MC.The lattice constants of 4H-SiC are:a=b=3.0780 °A,c=10.0460 °A andα=β=90°,γ=120°.
Selection of exchange-correlation functional can influenc the accuracy of calculation results.Therefore,the lattice constants and bulk modulus (B0) of Mg and SiC crystals were calculated using different exchangecorrelation functionals.Computed lattice constants andB0are listed in Table 1.Lattice constants of Mg using GGA-PBE functional area=b=3.2222 °A,c=5.1707 °A.The calculated results are very close to the experimental values,and also in well agreement with the values in Ref.[22] (a=b=3.222 °A,c=5.170 °A).The calculatedB0is 35.49 GPa,which is slightly lower than that in Ref.[22] (B0=36.3 GPa).Lattice constants using GGA-PW91 are close to that using GGA-PBE,but value ofB0is much lower.Lattice constants using LDA-CAPZ functional are lower (a=b=3.1597 °A,c=5.0766 °A) than that using GGA.The value ofB0(30.87 GPa) is also lower.Lattice constants(a=b=2.9390 °A,c=4.5208 °A) andB0(21.61 GPa) using HSE functional are all the lowest,and also much lower than the experimental values.As to the result of 4H-SiC,the calculated results with GGA-PBE and GGA-PW91 are close to the results in experiments and Ref.[31].Value of lattice constants (a=b=3.0553 °A,c=9.9649 °A) are much lower using HSE functional.In contrasting,value ofB0(224.13 GPa) is the highest,which is also higher than that in Ref.[21].Lattice constants (a=b=3.0414 °A,c=9.9546 °A)andB0(195.17 GPa) using LDA-CAPZ functional are all the lowest.Therefore,functional of GGA-PBE is used in the following calculations to ensure the accuracy.
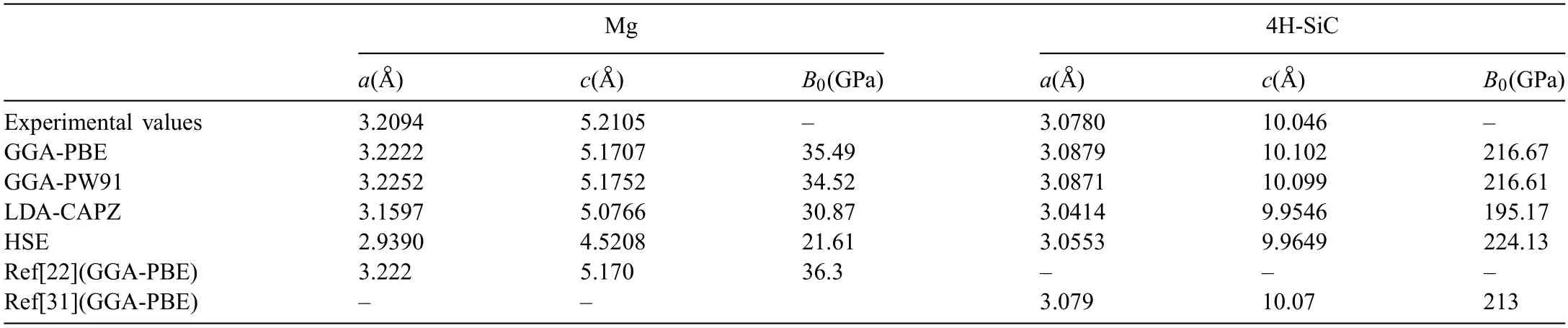
Table 1 Computed values of lattice constants and bulk modulus (B0) using different exchange-correlation functional.
3.2.Surface properties of Mg (0001) and 4H-SiC (0001)
4H-SiC (0001) polar surfaces are adopted for they can be either C-terminated (C-T) or Si-terminated (Si-T).The lattice constants of the SiC (0001) surface are:a=b=3.0780 °A andγ=120°.Mg (0001) surface is the closed-packed surface,and the lattice constants of Mg (0001) surface slab are:a=b=3.2094 °A andγ=120°.If Mg (0001) and SiC (0001)surfaces are matched together to form an interface,the misfi of interface is as low as 4.09%.
Firstly,Mg(0001)and SiC(0001)surface slabs were built.For the purpose of building thick enough slab to present bulklike properties,surface slabs with different thickness experienced fully relaxation.3~15 atomic layers of Mg(0001) and SiC(0001) surface slabs were built.Vacuum layer with thickness of 16 °A was added for each surface slab to eliminate the interactions between the surface atoms.For the SiC(0001)surfaces,both C-T and Si-T surfaces were considered.Changes of spacing (Δij) between neighboring layers were monitored,which can be calculated by Eq.(1):

Where dijand dij0are the interlayer distances of neighboring layers after and before geometry optimization,respectively.
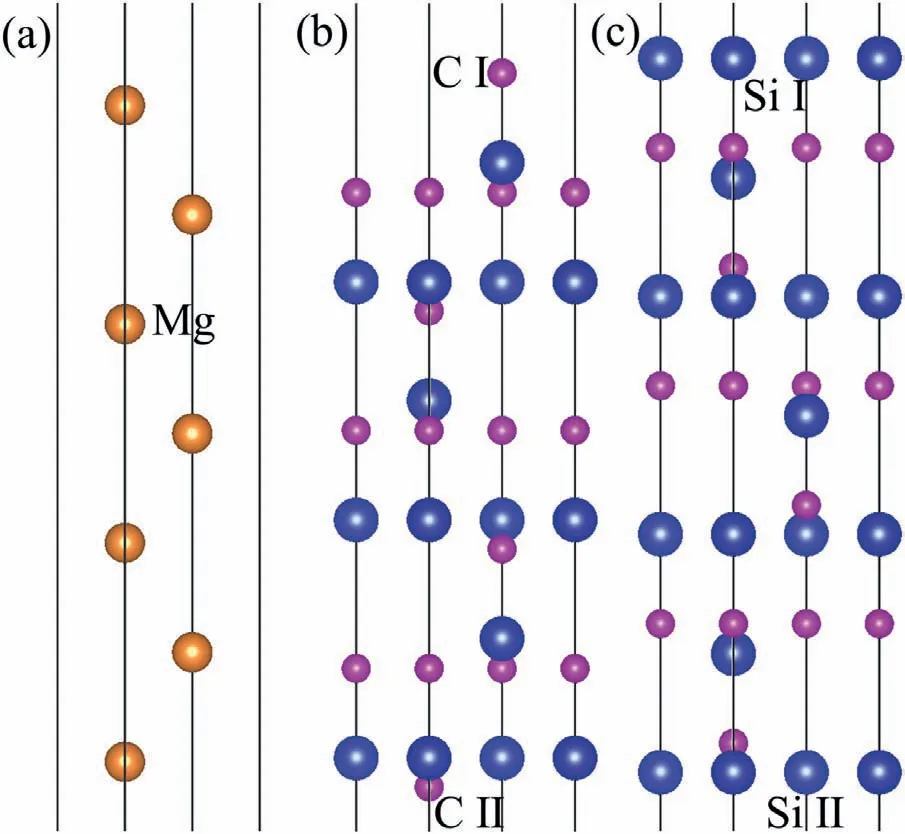
Fig.3.Surface configuration (a) Mg(0001),(b) C-T SiC(0001),(c) Si-T SiC(0001).
Results ofΔijafter fully relaxation are listed in Table.2.In general,value ofΔijdecreases with increase of thickness.For the Mg(0001) slabs,large values ofΔijare mainly found between the upper three layers.Δijbecomes relatively low when the slab is thicker than 5 atomic layers.For the CT SiC(0001) surface slab,value ofΔijbetween the second and third layers is always the largest,and it becomes very low when the slabs are thicker than 11 atomic layer.As to the Si-T SiC(0001) surface slab,much larger values ofΔijare found between the upper four atomic layers,and they decrease to lower values when the slab is thicker than 11 atomic layers.To ensure the calculation accuracy,7 atomiclayer Mg(0001) and 13 atomic-layer SiC (0001) surface slabs were built for both C-T and Si-T surfaces.The 13 atomiclayer 4H-SiC (0001) surfaces are non-stoichiometric,which can eliminate the dipole effect on the surfaces.C and Si atoms in the SiC slab have different positions.Therefore,C-T slabs have both C Ⅰ-T and C Ⅱ-T surfaces,Si-T slabs have both SiⅠ-T and Si Ⅱ-T surfaces,which are shown in Fig.3a),b) and c).
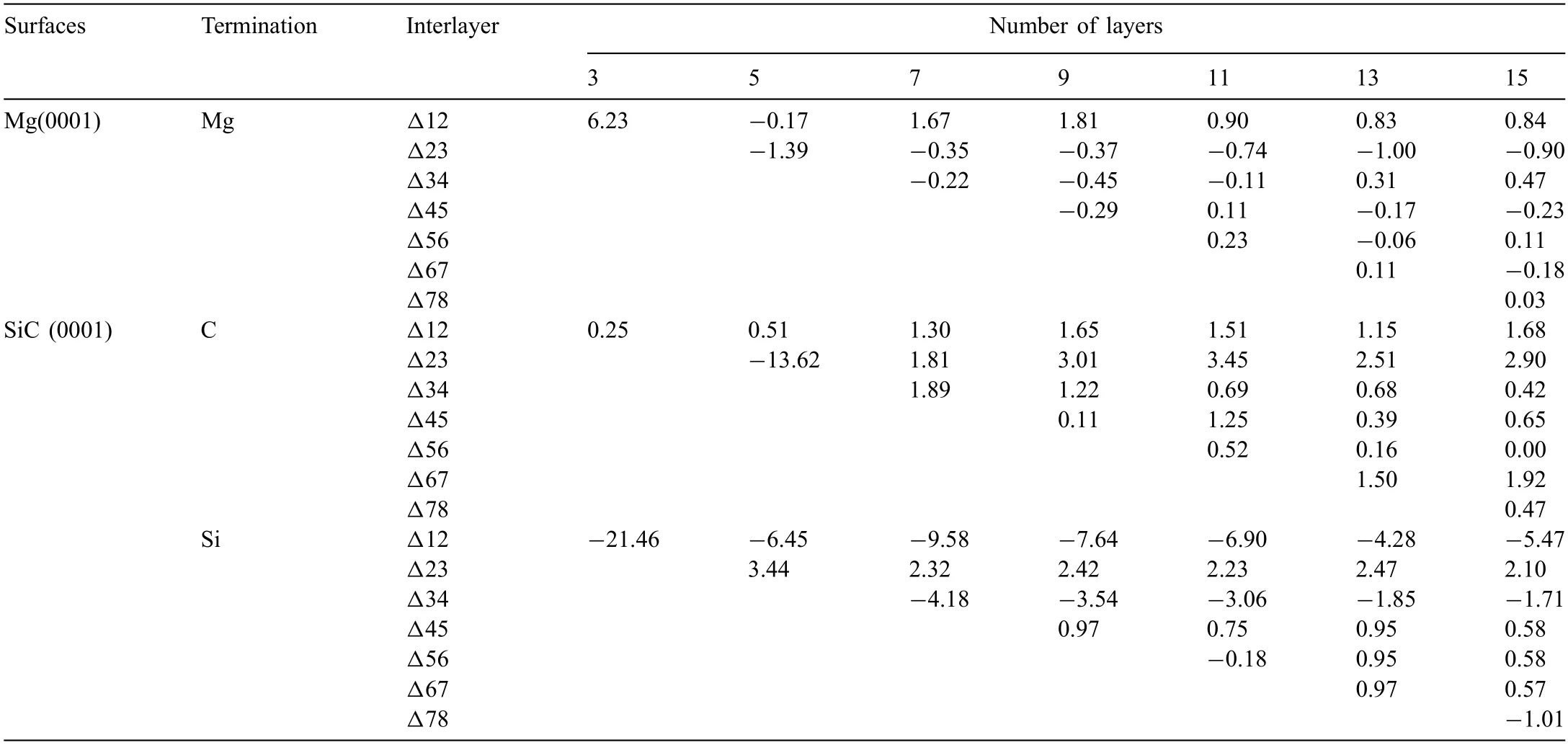
Table 2 Structure relaxation of Mg(0001) and SiC (0001) surfaces.
Surface energies of Mg(0001) and SiC (0001) surfaces were calculated.For Mg(0001) slab,surface energy (γ) can be calculated by Eq.(2) [32]:

WhereEslabandΔEare the energies of surface slab and single one atomic layer,Nis the atomic layers andArepresents the area of surface.Calculatedγof Mg(0001) is 0.52 J/m2.Surface energy of SiC (0001) is calculated considering chemical potential (μ) of both Si and C atoms.γcan be calculated by using Eq.(3) [33]:
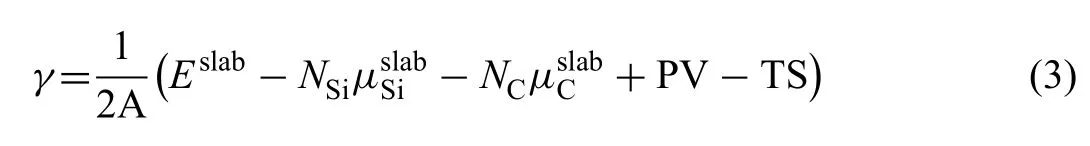
Where,Eslabis the total energy of the surface slab,Niandμiare numbers and chemical potentials of the corresponding atoms.Ais the area of surface slab.In general,the terms of PV and TS can be neglected at 0 K.
The total energy of the bulk SiCcan be given by Eqs.(4) and (5) [34]:
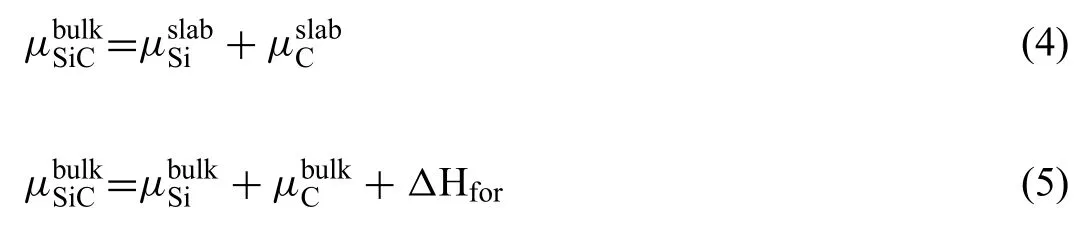
Where,are the chemical potentials of atoms in the corresponding slab and bulk.ΔHforis the formation enthalpy of the SiC.Value ofΔHforcan be calculated by Eq.(6):
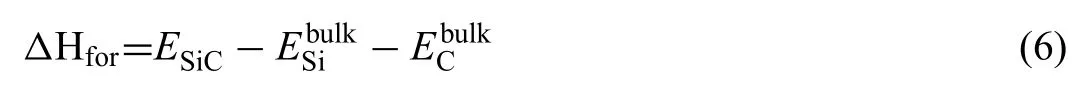
The chemical potential of atom in the slab is always lower than that in the bulk,therefore:


Fig.4.Relationship between surface energy of SiC(0001)slabs and chemical potential of C.
Surface energy of SiC can be calculated by Eq.(9):
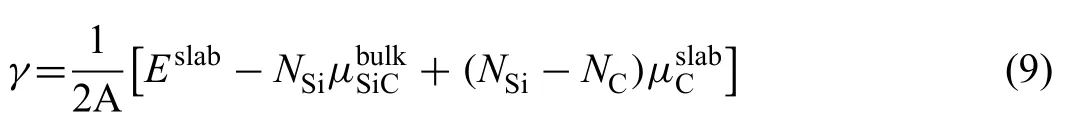
The calculated formation enthalpy of the SiC is 0.68 eV,which is very close to the result (0.66 eV) in Ref.[35].Fig.4 shows the relationship between surface energy of SiC(0001)surface and chemical potential of C.The surface energy is not a constant for the surface slab is either Si-rich or Crich.As increase of chemical potential of C,surface energy of C-T surface decreases from 8.59 J/m2to 7.92 J/m2,and surface energy of Si-T surface increases from 2.86 J/m2to 3.52 J/m2.It is found that surface energy of C-T slab is always larger than that of Si-T slab.The result indicates that C-T surface is more active.The termination of SiC(0001) surface can influenc the Mg/SiC interfacial bonding.
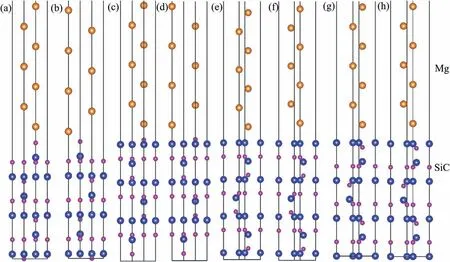
Fig.5.Interface configurations (a)C Ⅰ-T C Ⅰtop,(b) C Ⅰ-T C Ⅱtop,(c) C Ⅱ-T C Ⅱtop,(d) C Ⅱ-T Si Ⅱtop,(e) Si Ⅰ-T Si Ⅱtop,(f) Si Ⅰ-T Si Ⅰtop,(g) SiⅡ-T C Ⅱtop,(h) Si Ⅱ-T Si Ⅱtop.
3.3.Mg(0001)/SiC(0001) interfaces
Mg(0001)/SiC(0001) interfaces with different terminations and stacking sequences were constructed.When the SiC(0001) surfaces are terminated with C Ⅰ,C Ⅱ,Si Ⅰor Si Ⅱatom,the interfaces are named as CⅠ-T,CⅡ-T,SiⅠ-T or Si Ⅱ-T interfaces.For each interface,two kinds of stacking sequences were considered.When the Mg atom was placed on the top site of C Ⅰ,C Ⅱ,Si Ⅰor Si Ⅱatom directly,the interfaces are named as C Ⅰtop,C Ⅱtop,Si Ⅰtop or SiⅡtop interfaces.Therefore,8 kinds of interfaces were built,as shown in Fig.5a)~h).When building interface structure,optimized lattice parameters were used.The softer Mg(0001)slab was strained to match the SiC(0001) slab.For all the interfaces,a 16 °A-thick vacuum layer was added to separate the two free surfaces.
Work of adhesion (Wad) is used to evaluate the interfacial bonding strength quantitatively.Value of Wadcan be calculated by using Eq.(10) [36]:

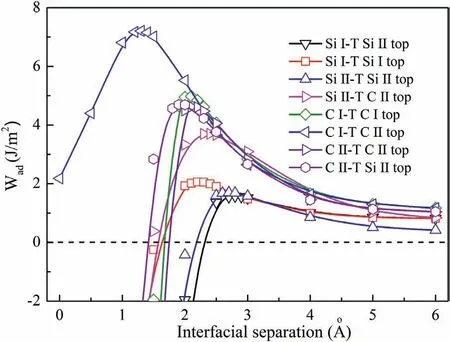
Fig.6.Relationship between Wad and interfacial separation.
Interfacial distance (d) represents vertical distance between the two slabs.In the calculating process,value of d was adjusted manually from 0 °A to 6 °A,and the corresponding Wadwas calculated.Results of Wadwith various d are shown in Fig.6.The curves all have the same trends.Peak value of Wadcan be found with the equilibrium interfacial distance(d0).In general,Wadof C-T interface is larger than that of Si-T interface.d0of C-T interface is smaller.Smaller value of d0always indicates shorter interfacial bonding length and stronger bonding.For the Si-T interfaces,Si Ⅱ-T C Ⅱtop interface has lowest Wadof 3.70 J/m2and d0of 2.4 °A.As to the C-T interfaces,Wadof C Ⅱ-T C Ⅱtop interface is 7.21 J/m2,which is much larger than that of other two interfaces.d0of C Ⅱ-T C Ⅱtop interface is only 1.3 °A,which is also the smallest in all interfaces.Results of Wadand d0imply that Mg-C bonds are stronger than Mg-Si bonds at the interface.Si Ⅱ-T C Ⅱtop and C Ⅱ-T C Ⅱtop interfaces with largest Wadare used to study the influenc of alloy elements in the following.
Fig.7.Side views of clean and doped interfaces after relaxation.(a) clean Si-T interface,(b) Si-T interface with Al,(c) Si-T interface with Zn,(d) Si-T interface with Zr,(e) clean C-T interface,(f) C-T interface with Al,(g) C-T interface with Zn,(h) C-T interface with Zr.
3.4.Mg(0001)/SiC(0001) interfaces with alloy elements
Al,Zn and Zr atoms are the common used alloy elements in AZ and ZK series Mg alloys and composites.Next,Al,Zn and Zr atoms were introduced to the clean Mg/SiC interfaces by replacing Mg atoms in-situ.In order to investigate the influenc of alloy elements coverage,2×2 supercells of Mg/SiC interfaces were built.Firstly,the selected clean Si-T and C-T interfaces were relaxed.Fig.7a) and e) show the Si-T and C-T interfaces after relaxation.Wadof Si-T and C-T interfaces are 3.76 J/m2and 7.65 J/m2after relaxation,which are both slightly higher than that before geometry optimization.Lengths of Mg-Si and Mg-C bonds at interfaces are 2.926 °A and 2.166 °A.Then one Al (or Zn,Zr) atom was doped into interface,and the interfaces also experienced relaxation.The relaxed Si-T and C-T interfaces with alloy elements are shown in Fig.7b)~d) and f)~h).During the relaxation process,Al dopant moved towards SiC slab at the Si-T interface.Bond length of Al-Si decreases to 2.898 °A after relaxation.While,Zn atom moved away from the SiC slab,and Zn-Si bond has length of 2.994 °A.It is also found that Zr and Si atoms moved towards each other and Zr-Si bond has length of 2.787 °A,which is much smaller than other bonds at interface.As to the doped C-T interfaces,Al,Zn and Zr atoms all moved closer to the C atoms.Bond lengths of Al-C,Zn-C and Zr-C bonds are shortened to 2.006 °A,2.005 °A and 2.082 °A,respectively.
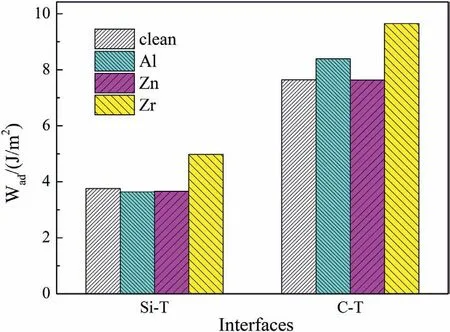
Fig.8.Wad of clean and doped Mg/SiC interfaces.
Fig.8 shows Wadvalues of clean and doped Mg/SiC interfaces.The clean Si-T interface has Wadof 3.76 J/m2.After doping Al and Zn atoms,values of Waddecrease to 3.63 J/m2and 3.66 J/m2.The results indicate that Al and Zn dopants would weaken the Si-T interfaces.When the interface is doped with Zr atom,Wadincreases to 4.97 J/m2.The large Wadand short Zr-Si bond length imply that Zr atom can form strong bond with Si atom,which can strengthen the Si-T interface.Wadof clean C-T interface is 7.65 J/m2.Zn dopant has little influenc on the bonding strength of C-T interface.Wadof Zn-doped C-T interface is 7.63 J/m2,which is almost the same as that of clean C-T interface.Al and Zr dopants can both improve Wadof C-T interfaces.Values of Wadincrease to 8.39 J/m2and 9.65 J/m2with Al and Zr atoms.Therefore,we can conclude that Zn atom can not improve interfacial bonding for both C-T and Si-T interfaces.Al atom can only strengthen the C-T interface.Zr atom can strengthen both C-T and Si-T interfaces.The strengthening effect of Zr atom is better than that of Al atom.
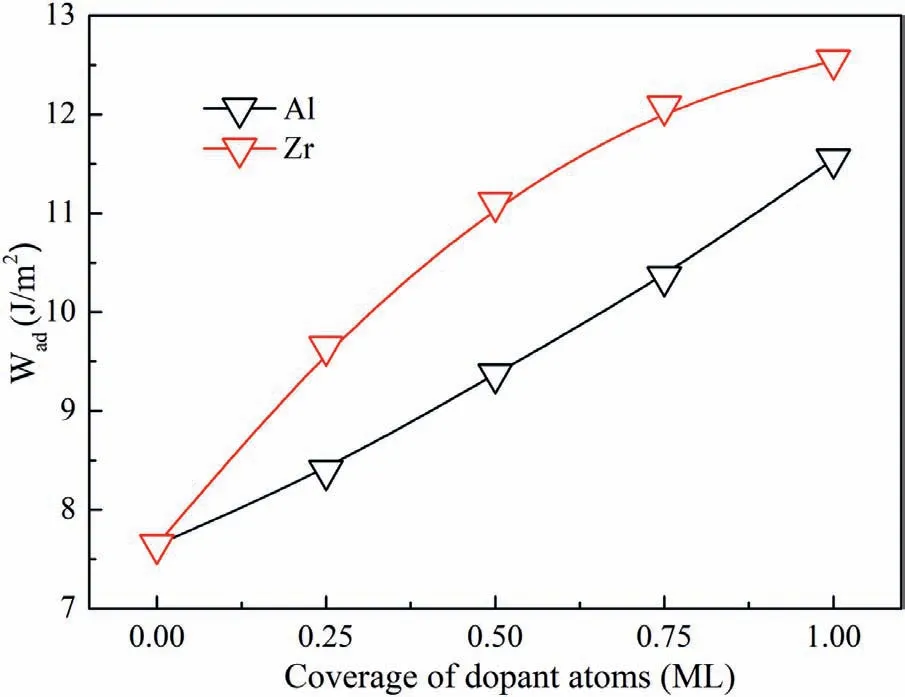
Fig.9.Values of Wad with different coverage of dopant atoms.
Different numbers of Al and Zr atoms were introduced into the C-T interfaces.Therefore,we can study the influenc of dopant numbers on the interfacial bonding.In the 2×2 C-T interface,each layer contains four atoms.Therefore,1,2,3 and 4 dopant atoms correspond to 0.25,0.5,0.75 and 1 monolayer (ML) of atom coverage.Fig.9 shows the values of Wadwith different ML of Al and Zr atoms.It is found that Wadincreases continually with coverage of dopant atoms.Wadof Zr-doped interface is always larger than that of Al-doped interface.When the interfaces are doped with four atoms,the interface will be saturated with Al-C and Zr-C bonds.Values of Wadincrease to 11.55 J/m2and 12.55 J/m2with 1 ML of Al and Zr atoms,which are much larger than that of clean Mg/SiC interfaces.
Heat of segregation (ΔGseg) is calculated to evaluate the stability of dopant atoms at the interfaces.The value ofΔGsegcan be obtained by Eq.(11) [37]:
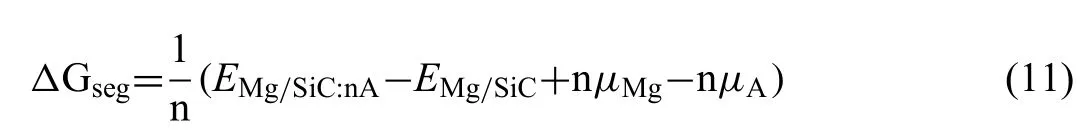
WhereEMg/SiC:nAandEMg/SiCrepresent total energies of the interfaces with dopant atoms and clean Mg/SiC interface.μand n are chemical potential and number of Al or Zr atoms.It is reported that more negative value ofΔGsegrepresents more stability in thermodynamics [38].
Fig.10 showsΔGsegvalues with different coverage of dopant atoms.When C-T interfaces are doped with one Al and Zr atom,ΔGsegare ?2.02 eV and ?3.98 eV.The negative values indicate that doping Al and Zr atoms to the CT Mg/SiC interface is stable in thermodynamics.The lowerΔGsegimplies that doping Zr atom is more stable.ΔGsegdecreases with increase of Al atom coverage,and increases with Zr atom coverage.Values ofΔGsegare all negative with different atom coverage.When the C-T interfaces are doped with 1 ML of Al and Zr atoms,ΔGsegare ?2.22 eV and?3.17 eV.The results indicate that the interfaces are still stable with four doped Al and Zr atoms.
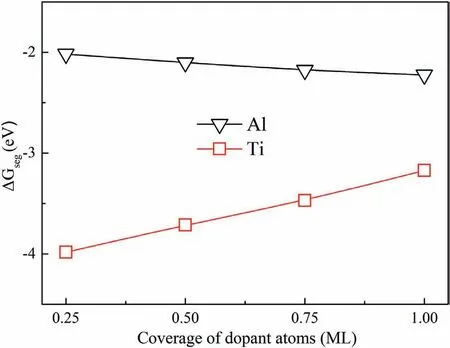
Fig.10.Relationships between ΔGseg and coverage of dopant atoms.
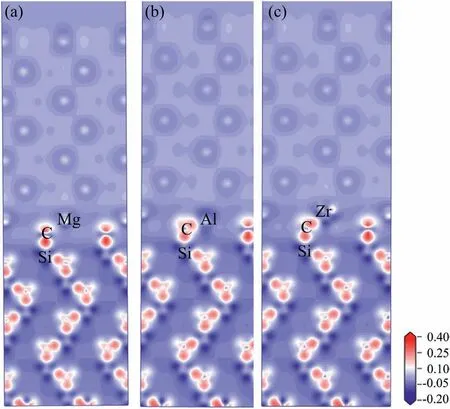
Fig.11.Charge density difference of Mg/SiC interfaces (a) clean interface,(b) Al-doped interface,(c) Zr-doped interface.
3.5.Electronic structures
To have a deeper insight on the bonding mechanism of the doped interfaces,electronic structures are analyzed through methods of charge density difference and partial density of states(PDOS).Results of charge density difference are shown in Fig.11.In the plots,the charge enrichment area is labeled as red color and charge dissipation area is labeled as blue color.Fig.11a) shows plot of charge density difference for clean interface.Red and blue areas are found around C and Mg atoms,which indicates that some electrons are transferred from Mg to C atoms.Mg and C atoms become cation and anion.They bond with each other through electrostatic attraction,which implies ionic bond.Fig.11b) and c) show charge density difference plots of Al and Zr-doped interfaces.Charge transfers are all found from the Al and Zr atoms to the neighboring C atoms.Therefore,Mg-C,Al-C and Zr-C bonds at the interface all have compositions of ionic bonds.Al-C and Zr-C bonds are stronger than the Mg-C bond for more transferred charges.
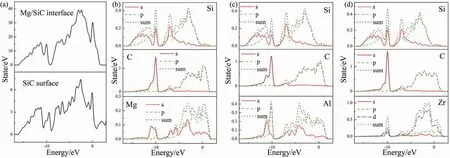
Fig.12.(a) totals DOS results of SiC and Mg/SiC interface,PDOS results of (b) clean Mg/SiC interface,(c) Al-doped interface,(d) Zr-doped interface.
Total DOS results of SiC and Mg/SiC interface are shown in Fig.12a).PDOS results of selected atoms in clean and doped Mg/SiC interface are shown in Fig.12b)~d).In the clean interface (Fig.12b),Mg-spand C-sporbitals overlap from ?9 eV to 1 eV.Overlapping of Mg-spand C-sporbital is also found within the energy range of ?12~?9.7 eV.These peaks are narrow,which represents localization of valence electrons.This kind of overlapping indicates covalent bond.Therefore,Mg-C bond at the interface is a mix of covalent and ionic bond.Bond population analysis is also performed to evaluate the degree of covalent.Bond population of Mg-C is 0.20,indicating that Mg-C bond is mainly ionic.Fig.12c)and d) show PDOS of atoms at Al and Zr-doped interfaces.The similar overlapping between Al(or Zr)and neighboring C atom is found,which indicates that Al-C and Zr-C bonds also have both ionic and covalent compositions.Bond populations of Al-C and Zr-C bonds are 0.93 and 1.02,which suggests that these two bonds are mainly covalent.
3.6.Discussion
SiC exists as form of solid particles in SiC/Mg composites for SiC has high melting point and high stability.Mg and 4H-SiC crystals all have hexagonal structure,and the lattice mismatch of Mg(0001)/SiC(0001) is only 4.09%.Therefore,SiC can be heterogeneity nucleus for Mg.When preparing SiC/Mg composites,wetting of Mg melt on SiC particles is the key factor to influenc the quality and properties of composites.Doping of Al and Zr atoms to the interface can raise Wadvalue,which also has influenc on wetting.The relationship between Wadand contact angle (θ) can be expressed as[39]:

WhereγLVis liquid-vapor interfacial tension.
When preparing SiC/Mg composites,γLVof Mg-Al (or Mg-Zr) alloy/SiC interface is a constant.Al and Zr atoms would diffuse to the interface and lead to larger Wad.The contact angle will be smaller with larger Wad.Therefore,segregation of Al and Zr to the interfaces can promote wetting and nucleation.
The calculation results are compared with the experimental results in the references.In Ref.[40],SiCw/AZ91 composites were prepared by squeeze cast at 800 °C.Massive Mg17Al12intermetallic compounds (IMC) are found at the interface between Mg alloy and SiC whiskers.The results confir segregation of Al to the Mg/SiC interface.Al would stay at the interface as form of Mg17Al12IMC.It also suggests that the IMC preferentially nucleates at the matrix-whisker interface for the strong bonds between Al and C atoms.In Ref.[41],SiC-reinforced magnesium alloy metal matrix composites are produced by rheocasting process.STEM micrograph as well as STEM/EDX maps are carried out to analyze the interface of SiC and Mg alloys.A lot of particles were found close to the SiC/metal interface,and the particles were rich in Al and C.Composition analysis shows the stoichiometry corresponds to AlC2.The results confir the strong bonds between Al and C,and Al-C compounds can form under certain conditions.In Ref.[42],SiCw/ZK51A magnesium matrix composite was fabricated by two-step squeeze casting.Through the TEM observation,SiCw/Mg interface was found to be quite clean,indicating no IMC formed during the fabrication process of the composite.It is also mentioned the SiC whiskers are very stable and carbides are difficul to form at the interface.From the results,we notice that although Zr can form strong bonds with C atoms,no Zr-C compounds formed at the interface.Mg alloys containing Zr can be excellent candidates for the matrix of composites.
4.Conclusions
In this paper,effects of Al,Zn and Zr atoms on the Mg(0001)/SiC(0001) interfacial bonding properties were investigated via methods of first-principl calculations.The C-T interfaces were found to have larger Wadthan the Si-T interfaces.C Ⅱ-T C Ⅱtop interface has largest Wadof 7.21 J/m2.Then Al,Zn and Zr atoms were doped into the Mg/SiC interfaces.Zn atom can not improve interfacial bonding for both C-T and Si-T interfaces.Al atom can only strengthen the C-T interface.Zr atom can strengthen both C-T and Si-T interfaces.Wadof Zr-doped C-T interface is improved to 9.65 J/m2.In the Al and Zr-doped C-T interfaces,Wadincreases as the ML of atom coverage.Wadcan reach up to 11.55 J/m2and 12.55 J/m2with 1 ML of Al and Zr atoms.Increase of Wadcan lead to lower contact angles,which can improve wetting and nucleation.NegativeΔGsegvalues imply that doping Al and Zr to the C-T interface is stable in thermodynamics.Al-C and Zr-C bonds formed at interfaces have composition of both ionic and covalent.Al-C and Zr-C bonds have more covalent composition than the Mg-C bond,which is responsible for strong bonding strength.Experimental results in references were also analyzed,which can confir the calculation results.
Conflic of Interest
None.
Acknowledgments
This research was funded by China Postdoctoral Science Foundation (No.2020M670670),Foundation Strengthening Program (No:2019-JCJQ-00),Natural Science Foundation of Hebei Province (E2019202407),Science and Technology Research Project of Hebei Province Colleges and Universities(QN2019028) and“Yuan Guang Scholar”Plan of Hebei University of Technology.
 Journal of Magnesium and Alloys2022年6期
Journal of Magnesium and Alloys2022年6期
- Journal of Magnesium and Alloys的其它文章
- EDITORIAL BOARD
- Aims and Scope
- Surface oxidation study of molten Mg-Al alloys by oxide/metal/oxide sandwich method
- Production and characterisation of new bioresorbable radiopaque Mg-Zn-Y alloy to improve X-ray visibility of polymeric scaffolds
- Quantitative study on the tension-compression yield asymmetry of a Mg-3Al-1Zn alloy with bimodal texture components
- Microstructure analyses and phase-fiel simulation of partially divorced eutectic solidificatio in hypoeutectic Mg-Al Alloys